Natalizumab: Therapieoption bei hochaktiver RRMS
Interessengebiete: Allgemeinmedizin und Innere Medizin, Neurologie, Biosimilars
Die Multiple Sklerose (MS) ist mit etwa 280.000 Betroffenen die häufigste chronisch-entzündliche Erkrankung des Zentralnervensystems (ZNS) in Deutschland – Tendenz steigend. Die Erkrankung wird durch autoimmun vermittelte, inflammatorische Prozesse verursacht, die zu Demyelinisierung von Axonen, ZNS-Läsionen und Neurodegeneration führen.
Durch den Einsatz moderner verlaufsmodifizierender Medikamente und eine frühere Diagnosestellung hat sich die Prognose der MS in den letzten Jahrzehnten deutlich verbessert. Einer der bewährten und hochpotenten Wirkstoffe zur verlaufsmodifizierenden Therapie der MS ist Natalizumab: Der 2006 in der EU zugelassene Wirkstoff war der erste monoklonale Antikörper zur Behandlung der hochaktiven, schubförmig remittierenden MS (RRMS). Natalizumab gehört zu den verlaufsmodifizierenden Therapeutika der Wirksamkeitskategorie 3 gemäß S2k-Leitlinie und kann die Krankheitsaktivität und die Zahl der entzündlichen Läsionen im Vergleich zu niedrigpotenten Wirkstoffen signifikant reduzieren.
In der vorliegenden Fortbildung werden grundlegende Kenntnisse zu Natalizumab vermittelt. Nach einer allgemeinen Einleitung zur Erkrankung (Teil 1) und zu Grundlagen des Wirkstoffs sowie biologischen Arzneimitteln im Allgemeinen (Teil 2) werden im weiteren Verlauf die Besonderheiten der Therapie (Teil 3) erörtert. Im vierten Teil der Fortbildung werden die Studiendaten zur Wirksamkeit und Sicherheit vom Referenz-Natalizumab und seinem Biosimilar erläutert.
Kursinhalt
Inhaltsverzeichnis
Einleitung und Zielsetzung
Durch den Einsatz moderner verlaufsmodifizierender Medikamente und eine frühere Diagnosestellung hat sich die Prognose der Multiplen Sklerose (MS) in den letzten Jahrzehnten signifikant verbessert. Die zugelassenen Immuntherapeutika können durch ihre antiinflammatorische Wirkung die Krankheitsaktivität reduzieren und die Progression der Erkrankung verzögern bzw. verhindern.1,2
Einer der bewährten und hochpotenten Wirkstoffe zur verlaufsmodifizierenden Therapie der MS ist Natalizumab: Der im Jahre 2006 in der EU zugelassene Wirkstoff war der erste monoklonale Antikörper zur Behandlung der hochaktiven, schubförmig remittierenden MS (RRMS).3 Natalizumab gehört zu den verlaufsmodifizierenden Therapeutika der Wirksamkeitskategorie 3 gemäß S2k-Leitlinie und kann die Krankheitsaktivität und die Zahl der entzündlichen Läsionen im Vergleich zu niedrigpotenten Wirkstoffen signifikant reduzieren.4
In der vorliegenden Fortbildung werden grundlegende Kenntnisse zu Natalizumab vermittelt. Nach einer allgemeinen Einleitung zur Erkrankung (Teil 1) und zu Grundlagen des Wirkstoffs sowie biologischen Arzneimitteln im Allgemeinen (Teil 2) werden im weiteren Verlauf die Besonderheiten der Therapie (Teil 3) erörtert. Im vierten Teil der Fortbildung werden die Studiendaten zur Wirksamkeit und Sicherheit vom Referenz-Natalizumab und seinem Biosimilar erläutert.
1 Multiple Sklerose
Die Multiple Sklerose (MS) ist mit etwa 280.000 Betroffenen die häufigste chronisch-entzündliche Erkrankung des Zentralnervensystems (ZNS) in Deutschland – Tendenz steigend. Die Erkrankung wird durch autoimmun vermittelte, inflammatorische Prozesse verursacht, die zur Demyelinisierung von Axonen, ZNS-Läsionen und Neurodegeneration führen. Das bessere Verständnis der zugrundeliegenden Pathomechanismen hat in den letzten Jahrzehnten die Entwicklung neuer verlaufsmodifizierender Immuntherapeutika vorangetrieben, die die Therapieoptionen wesentlich erweitern.4 Derzeit beschäftigt sich die MS-Forschung mit verschiedenen Fragestellungen, etwa nach der optimalen Abfolge der verschiedenen Therapeutika oder etwaigen Deeskalationsstrategien. Zudem werden auch neue Wirkstoffkandidaten, wie Bruton-Tyrosinkinase(BTK)-Inhibitoren, in klinischen Studien auf ihre Wirksamkeit und Sicherheit hin evaluiert.1,5 Die Prognose von MS-Betroffenen hat sich in den letzten Jahrzehnten wesentlich verbessert, dies ist auf verschiedene Faktoren wie eine frühzeitigere Diagnosestellung durch veränderte Diagnosekriterien,
z. B. beim klinisch isolierten Syndrom (KIS), oder die erweiterten Therapiemöglichkeiten zurückzuführen.1,2
Insbesondere den immunmodulatorischen Wirkstoffen kommt in der modernen MS-Therapie eine zentrale Rolle zu: Durch gezielte Prävention entzündlicher Läsionen können sie die Progression der Erkrankung hinauszögern.4,6 Die Immuntherapeutika werden dabei sowohl zur Behandlung der schubförmig remittierenden MS (RRMS) als auch der progredienten Verlaufsformen, der primär progredienten MS (PPMS) und der sekundär progredienten MS (SPMS), eingesetzt.
Gemäß der Leitlinie der Deutschen Gesellschaft für Neurologie werden die gegenwärtig verfügbaren Immuntherapeutika entsprechend ihrer Wirkstärke – sprich dem Potenzial zur Reduktion der Schubrate im Vergleich zu Placebo – in drei Wirksamkeitskategorien unterteilt (Tab. 1).4 Bei der Wahl der Therapie sollte individuell der potenzielle Nutzen der Therapie gegen mögliche Risiken abgewogen werden, wobei verschiedene Faktoren wie Verlaufsform, Krankheitsaktivität, Patientenpräferenzen (Applikationsart, Therapieintervalle, Familienplanung), Komorbiditäten und das Nebenwirkungsprofil des Arzneimittels in die Entscheidungsfindung mit einfließen.4
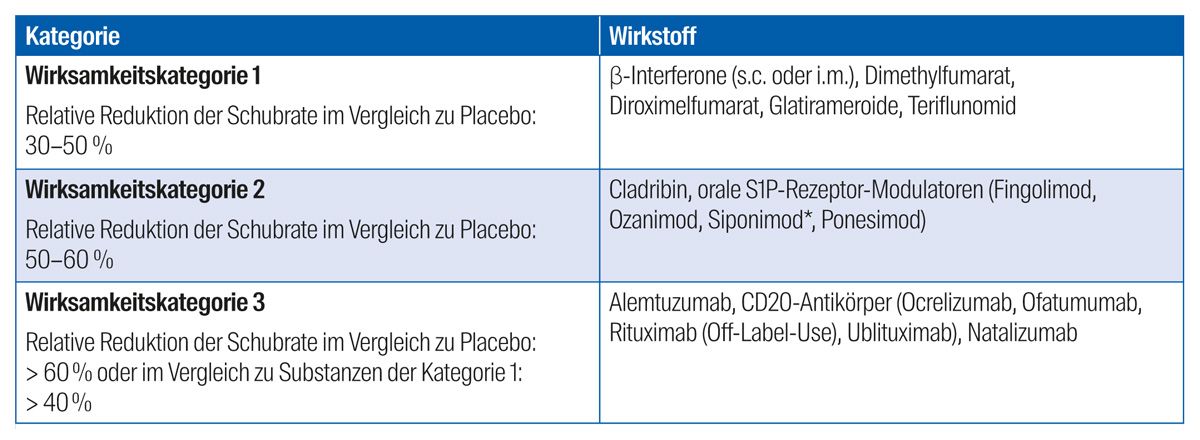
Tab. 1: Einteilung der Immuntherapeutika
2 Grundlagen zu Natalizumab
2.1 Struktur und Wirkmechanismus
Natalizumab gehört zu den biologischen Arzneimitteln (Biologika) und wird in murinen Zelllinien bzw. in der Chinese-Hamster-Ovary(CHO)-Zelllinie mithilfe von rekombinanter DNA-Technologie hergestellt (siehe Kasten).3 Bei Natalizumab handelt es sich um einen rekombinanten, humanisierten monoklonalen Antikörper.3
Natalizumab bindet an die α4-Untereinheit der humanen Integrine α4β1 und α4β7. Diese werden auf der Oberfläche von Leukozyten exprimiert.3 Integrine spielen eine wichtige Rolle für die Zelladhäsion und die Rekrutierung von Immunzellen ins Gewebe (engl. homing). Die spezifische Bindung von Natalizumab an α4β1- bzw. α4β7-Integrine blockiert die Wechselwirkung dieser mit den entsprechenden Rezeptoren vascular cell adhesion molecule-1 (VCAM-1) und mucosal addressing cell adhesion molecule-1 (MadCAM-1), die auf der Zellmembran von Endothelzellen exprimiert werden. Dadurch wird die transendotheliale Migration von mononukleären Leukozyten über die Blut-Hirn-Schranke in entzündetes Parenchymgewebe verhindert, wodurch das inflammatorische Geschehen im Zentralnervensystem (ZNS) reduziert wird (Abb. 1).3
Möglicherweise hemmt Natalizumab auch bereits bestehende Entzündungen im betroffenen Gewebe durch Inhibition der Bindung von α4-exprimierenden Leukozyten an ihre Liganden in der extrazellulären Matrix.3
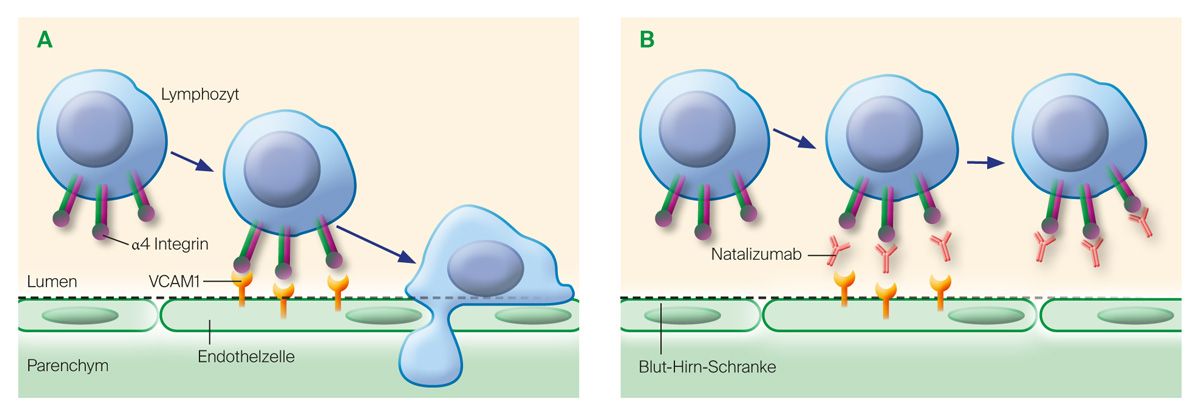
Abb. 1: Natalizumab hemmt die transendotheliale Migration von mononukleären Lymphozyten; modifiziert nach Steinman
Exkurs: Biologika
Biologika weisen im Vergleich zu klassischen, chemisch synthetisierten Wirkstoffen einige Besonderheiten auf. Die folgenden Besonderheiten gelten sowohl für Referenzarzneimittel als auch für Biosimilars.
Größe und Komplexität
Bei Biologika handelt es sich i. d. R. um Proteine, die im Vergleich zu den meisten chemisch synthetisierten Wirkstoffen deutlich größer und komplexer sind. Ihre charakteristische 3D-Struktur – bedingt durch die Aminosäuresequenz und damit die Faltung der Peptidketten – ist entscheidend für die Wirkungsweise.
Viele Biologika weisen zudem ein spezifisches Glykosylierungsmuster (Anheftung von Zuckermolekülen) auf.
Herstellungsverfahren und Mikroheterogenität
Die Herstellung im lebenden Expressionssystem (Zelllinien, Organismen) und die Komplexität der Wirkstoffe führt zu kleinen Variabilitäten zwischen verschiedenen Produktchargen eines Biologikums und wird als Mikroheterogenität bezeichnet. Aminosäuresequenz und 3D-Struktur müssen identisch sein, lediglich geringe Abweichungen bei posttranslationalen Modifikationen, wie z. B. Glykosylierung, sind zulässig, sofern diese die biologische Aktivität nicht verändern und klinisch nicht relevant – im Sinne von Wirksamkeit, Sicherheit und Immunogenität – sind. Durch ein streng reguliertes Herstellungsverfahren und umfangreiche Qualitätskontrollen wird sichergestellt, dass Biologika die bei der Zulassung definierten Wirkstoffspezifikationen langfristig einhalten.
Potenzielle Immunogenität
Biologische Arzneimittel haben das Potenzial, eine Immunreaktion auszulösen, was vorwiegend auf ihre Größe und Komplexität zurückzuführen ist. Aus diesem Grund wird im Rahmen der Zulassung grundsätzlich die Immunogenität von Biologika untersucht und im weiteren Verlauf überwacht. Im Vordergrund der Immunreaktion stehen dabei gegen den Wirkstoff gerichtete Antikörper (engl. anti-drug antibodies, ADA). Diese können die Wirksamkeit und Sicherheit biologischer Arzneimittel beeinflussen.
2.2 Indikation, Anwendung und Dosierung
Indikation
Natalizumab ist gemäß der S2k-Leitlinie ein Wirkstoff der Wirksamkeitskategorie 3.4 Er ist zur Monotherapie bei Erwachsenen mit hochaktiver, schubförmig remittierender Multipler Sklerose (RRMS) zugelassen, bei denen
- trotz einer Behandlung mit einem vollständigen und angemessenen Zyklus mit mindestens einer krankheitsmodifizierenden Therapie (DMT) eine hochaktive Erkrankung vorliegt oder
- bei denen eine rasch fortschreitende RRMS vorliegt. Diese wird definiert durch 2 oder mehr Schübe mit Behinderungsprogression innerhalb eines Jahres und mit einer oder mehr Gadolinium-anreichernden Läsionen in der Magnetresonanztomographie (MRT) des Gehirns oder mit einer signifikanten Erhöhung der T2-Läsionen im Vergleich zu einer kürzlich durchgeführten MRT.3
Kontraindikationen
Natalizumab ist bei einigen Patientengruppen kontraindiziert:3
- Immungeschwächte Patienten mit einem erhöhten Risiko für opportunistische Infektionen
- Patienten mit progressiver multifokaler Leukenzephalopathie (PML)
- Patienten, die eine andere krankheitsmodifizierende Therapie erhalten
- Patienten mit Malignomen (Ausnahme Basaliom)
Wirksamkeit und Sicherheit von Natalizumab sind bei Kindern und Jugendlichen nicht erwiesen, und der Wirkstoff wird dementsprechend nicht bei dieser Patientengruppe eingesetzt. Bei älteren Patienten über 65 Jahre wird die Anwendung von Natalizumab nicht empfohlen, da zu dieser Gruppe keine Daten vorliegen.3
Anwendung und Dosierung
Die Therapie mit Natalizumab sollte nur durch einen erfahrenen Spezialisten erfolgen und ein rascher Zugang zu einer MRT muss gewährleistet sein. Natalizumab wird alle 4 Wochen verabreicht, wobei die empfohlene Dosis bei 300 mg liegt. Der Wirkstoff wird entweder als intravenöse Infusion (1-mal 300 mg) oder subkutan mittels Fertigspritze (2-mal 150 mg) verabreicht.3
3 Besonderheiten der Therapie
3.1 PML und Anti-JCV-Antikörper-Testung
Bei der Anwendung von Natalizumab besteht ein erhöhtes Risiko für die Entwicklung einer progressiven multifokalen Leukenzephalopathie (PML). So wurden während der Behandlung mit Natalizumab und bis zu 6 Monate nach der letzten Dosis Fälle von PML berichtet.3,9 Bei der PML handelt es sich um eine seltene opportunistische Infektion, die durch Reaktivierung des John-Cunningham-Virus (JCV) hervorgerufen wird und zu tödlichen Verläufen bzw. schweren Behinderungen führen kann (916 bestätige PML-Fälle bei MS unter Natalizumab-Therapie, Gesamtinzidenz: 34,8 pro 10.000 mit Natalizumab behandelte Patienten; Stand: 01.08.2023).3,4,10
Der Erstkontakt mit dem Virus findet mutmaßlich in der Kindheit statt, während das Virus im Erwachsenenalter hauptsächlich asymptomatisch in der Niere persistiert.10 Die Prävalenz von Anti-JCV-Antikörpern lag in einer Querschnittsstudie in der EU bei 48,8–69,5 %.11 Bei der PML infiziert das reaktivierte JCV Oligodendrozyten, was infolge zu Demyelinisierung und ZNS-Läsionen führt. Neben Oligodendrozyten können auch Körnerzellen im Kleinhirn vom JCV infiziert werden, dies löst eine JCV-Körnerzellen-Neuronopathie (engl. granule cell neuronopathy; GCN) aus; diese kann mit ähnlichen Symptomen wie eine PML einhergehen.12,13 Die klinische Manifestation einer PML ist – insbesondere zu Beginn – nur schwer von neurologischen Symptomen der MS zu unterscheiden, zudem sind auch asymptomatische PML-Fälle berichtet worden.14
Risikofaktoren und Anti-JCV-Antikörper-Testung
Folgende Faktoren sind mit einem erhöhten Risiko für die Entwicklung einer PML assoziiert und werden zur Risikostratifizierung bestimmt (Abb. 2):3,15
- Vorliegen von Anti-JCV-Antikörpern
- Behandlungsdauer mit Natalizumab > 2 Jahre
- Vorangegangene Behandlung mit Immunsuppressiva
Vor Beginn einer Therapie mit Natalizumab wird eine Testung auf Anti-JCV-Antikörper empfohlen, hierzu kommt ein ELISA (engl. enzyme-linked immunosorbent assay) zum Einsatz.16,17 Die Inzidenz der PML liegt bei negativem Antikörperstatus bei 0,1/1.000 mit Natalizumab behandelten Patienten.15 Die Testung auf Anti-JCV-Antikörper sollte bei testnegativen Patienten alle 6 Monate wiederholt werden, um falsch-negative Testergebnisse oder neu erworbene JCV-Infektionen zu identifizieren.3
Bei positivem Antikörperstatus ist das PML-Risiko erhöht – liegen alle 3 Risikofaktoren vor, ist das Risiko signifikant erhöht.15 Aus diesem Grund fließen diese Faktoren in den Algorithmus der Risikoabschätzung mit ein (Abb. 2): die Therapiedauer mit Natalizumab, eine mögliche Vorbehandlung mit Immunsuppressiva und der Anti-JCV-Antikörperindex (Antikörpertiter).16,17 Bei testpositiven Patienten, die nicht mit Immunsuppressiva vorbehandelt wurden, korreliert die Höhe des Anti-JCV-Antikörpertiters mit dem PML-Risiko. Bei einem Indexwert von ≤ 0,9 scheint das PML-Risiko niedrig zu sein, bei Werten ≥ 1,5 und einer 2-jährigen Behandlungsdauer mit Natalizumab steigt das Risiko erheblich an.15 Derzeit existieren zwei verschiedene Anti-JCV-Antikörpertests: Ein Vergleich zwischen den beiden Tests zeigt eine mögliche Abweichung von bis zu 0,1 bei den Indexwerten (z. B. unterer Bereich = niedriges Risiko: ≤ 0,8 bzw. oberer Bereich = hohes Risiko: ≥1,4).17
Es wird empfohlen, testnegative Personen alle 6 Monate einer Testung auf Anti-JCV-Antikörper zu unterziehen. Außerdem sollte der Antikörpertiter bei Patienten, die nicht mit Immunsuppressiva vorbehandelt wurden und die einen niedrigen Antikörpertiter aufweisen, nach 2-jähriger Behandlungsdauer mit Natalizumab erneut regelmäßig (alle 6 Monate) bestimmt werden.3
Eine Plasmapherese bzw. ein Plasmaaustausch oder die intravenöse Gabe von Immunglobulinen kann die Interpretation des Anti-JCV-Antikörper-Tests beeinflussen. Daher sollte 2 Wochen nach einer Plasmapherese bzw. einem Plasmaaustausch keine Antikörper-Testung erfolgen – bei einer intravenösen Immunglobulin-Gabe beträgt dieser Zeitraum 6 Monate.3
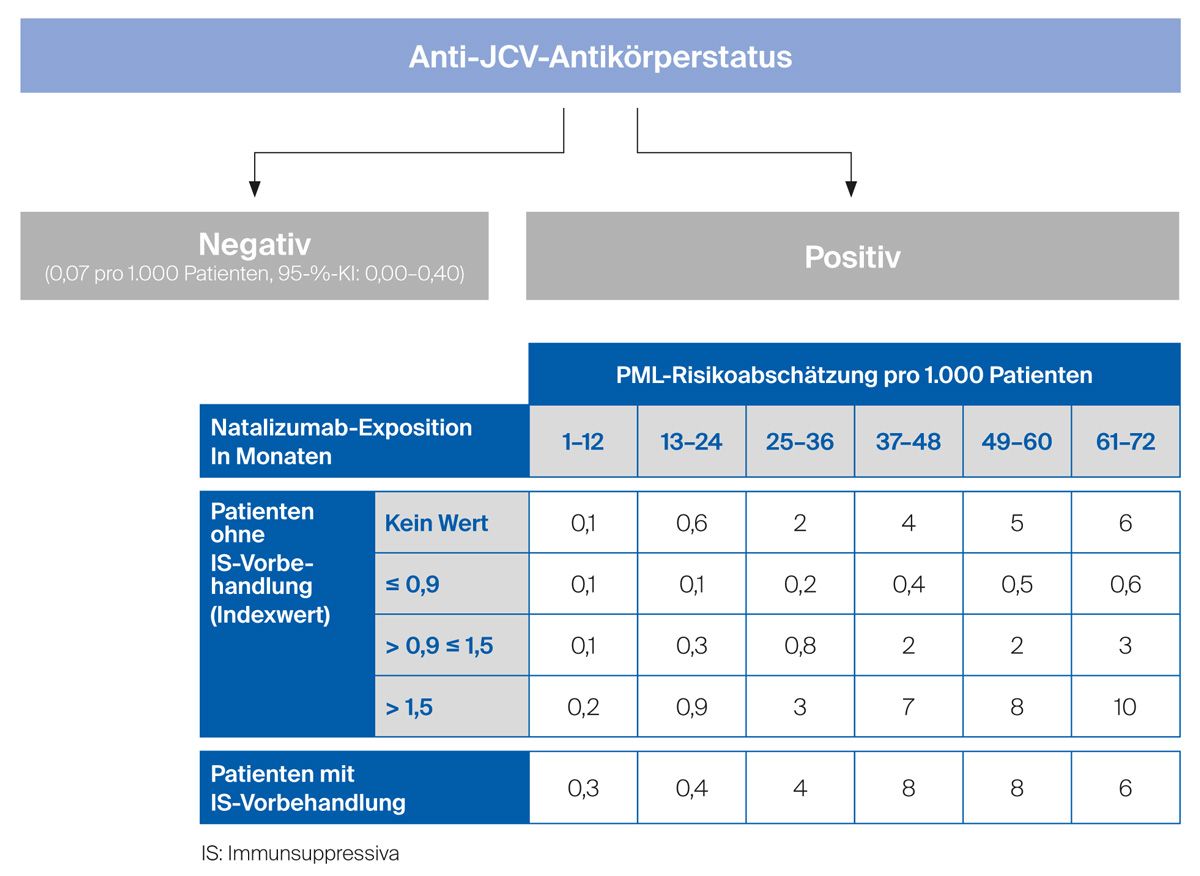
Abb. 2: Algorithmus der Risikoabschätzung zur Entwicklung einer PML. Daten nach Ho et al.
MRT-Untersuchungen
Vor Beginn der Therapie mit Natalizumab sollte eine MRT-Untersuchung zu Referenzzwecken erfolgen, die mindestens 1-mal jährlich wiederholt wird. Bei Patienten mit einem erhöhten PML-Risiko sollten kürzere Intervalle, z. B. im Abstand von 3 bis 6 Monaten, in Erwägung gezogen werden.3,18,19 Beim Auftreten neuer neurologischer Symptome muss eine PML bzw. JCV-GCN stets differentialdiagnostisch berücksichtigt werden. Bei Verdacht auf eine PML oder eine JCV-GCN muss die Therapie unterbrochen werden, bis diese ausgeschlossen werden können.3 Dazu entscheidet der behandelnde Arzt, ob die Symptome auf eine neurologische Dysfunktion hinweisen und ob diese MS-typisch sind oder auf eine PML bzw. JCV-GCN hindeuten. Sollten Zweifel bei der Entscheidung bestehen, werden MRT-Untersuchungen sowie Liquortests auf DNA des JCV durchgeführt. Bestätigt sich eine PML oder JCV-GCN, muss die Therapie dauerhaft abgesetzt werden. Wenn eine PML und/oder JCV-GCN jedoch ausgeschlossen wurden, kann die Therapie mit Natalizumab wieder aufgenommen werden.3
Zur PML-Risikoreduktion werden verschiedene Maßnahmen umgesetzt, die das Nutzen-Risiko-Verhältnis der Natalizumab-Therapie günstig beeinflussen sollen (siehe Kasten). So stehen beispielsweise umfangreiche Schulungsmaterialien zur Verfügung, die die behandelnden Ärzte über eine sichere und wirksame Anwendung von Natalizumab informieren.16,17
Maßnahmen zur Risikominimierung3,15-17
- PML-Risikostratifizierung vor Therapiebeginn
- Regelmäßige Anti-JCV-Antikörper-Testung
- Regelmäßige MRT-Kontrolluntersuchungen (bis 6 Monate nach Beendigung der Therapie)18,19
- Schulungsmaterial für Ärzte (erhöhte klinische Wachsamkeit)3,16,17
- Regelmäßige Aufklärung von Patienten, Angehörigen und Pflegepersonal (einschl. Aushändigung eines Patientenpasses,3,16,17 erneutes Aufklärungsgespräch nach 2-jähriger Behandlungsdauer oder bei Beenden der Behandlung)
- Therapieunterbrechung bei Verdacht auf PML oder andere opportunistische Infektion
3.2 Behandlungsdauer und besondere Patientengruppen
Natalizumab wird alle 4 Wochen – als intravenöse Infusion oder als subkutane Injektion – verabreicht. Sollten sich nach 6-monatiger Behandlungsdauer keine Hinweise auf einen Behandlungserfolg zeigen, ist ein Fortsetzen der Therapie sorgfältig zu hinterfragen.3 Klinische Studiendaten zur Sicherheit und Wirksamkeit von Natalizumab liegen für eine Behandlungsdauer von bis zu 2 Jahren vor, allerdings nur für die intravenöse Verabreichung.3 Nach 2-jähriger Behandlung sollte die Therapie nur dann fortgesetzt werden, wenn zuvor eine erneute Nutzen-Risiko-Abwägung stattgefunden hat. Darüber hinaus muss der Patient erneut über die Risikofaktoren für die Entwicklung einer PML, wie etwa eine Behandlungsdauer > 2 Jahre, informiert werden.3
Besondere Patientengruppen
Natalizumab sollte in der Schwangerschaft nur dann angewendet werden, wenn dies eindeutig erforderlich ist. In tierexperimentellen Studien zeigten sich reproduktionstoxische Effekte.3 Bei Frauen, die unter Natalizumab-Therapie schwanger werden, sollte ein Absetzen der Behandlung in Erwägung gezogen werden. Dabei ist der klinische Zustand der Patientin und ein mögliches Wiederauftreten der Krankheitsaktivität in die Nutzen-Risiko-Abwägung mit einzubeziehen.3 Aufgrund von Berichten über Thrombozytopenien und Anämien bei Säuglingen, deren Mütter mit Natalizumab während der Schwangerschaft behandelt wurden, sollten Neugeborene auf entsprechende hämatologische Parameter hin untersucht werden.3 In der Stillzeit soll die Behandlung mit Natalizumab unterbrochen werden, da der Wirkstoff in die Muttermilch übergeht.3
Es wurden keine Untersuchungen an Patienten mit Nieren- und Leberfunktionsstörungen durchgeführt, sodass keine Daten zur Pharmakokinetik des Wirkstoffs bei diesen Patienten vorliegen, eine Dosisanpassung ist aber vermutlich nicht notwendig.3
4 Daten zur Wirksamkeit und Sicherheit von Natalizumab
Die Wirksamkeit und Sicherheit von neuen Arzneimitteln muss für die Zulassung im Rahmen von klinischen Studien gezeigt werden. Bei den zulassungsrelevanten Studien handelt es sich i. d. R. um kontrollierte, randomisierte Phase-3-Studien. Bevor diese für Natalizumab vorgestellt werden, soll zunächst im Unterkapitel 4.1 auf das Zulassungsverfahren von Biologika (einschließlich Biosimilars) eingegangen werden, da es einige Besonderheiten aufweist.
4.1 Zulassungsverfahren und Qualitätsüberwachung von Biologika
Für die Zulassung von Biologika werden hohe Anforderungen an ihre Qualität gestellt. Zum einen muss die klinische Wirksamkeit und Sicherheit des Arzneimittels hinreichend belegt werden, zum anderen muss die Reproduzierbarkeit und Zuverlässigkeit des Herstellungsverfahrens dargelegt werden. Um zu gewährleisten, dass die den Biologika inhärente Variabilität keinen Einfluss auf die Wirksamkeit und Sicherheit des Arzneimittels hat, werden entsprechend bei der Zulassung und in der späteren Wirkstoffproduktion zwei wesentliche Aspekte berücksichtigt:20
- Festlegung der Wirkstoffspezifikationen bei der Zulassung von biologischen Arzneimitteln mithilfe verschiedener Analyseverfahren (Aminosäuresequenz, 3D-Struktur und biologische Aktivität).
- Bestimmung des Ausmaßes der erlaubten Mikroheterogenität für weitere Produktchargen. Es wird auch häufig vom „Korridor der erlaubten Mikroheterogenität“ gesprochen. Dieser gibt vor, welche Abweichungen im Glykosylierungsmuster zukünftig erlaubt sind, ohne dass Wirksamkeit und Sicherheit beeinflusst werden.
Aufbauend auf präklinischen Studien zur pharmazeutischen Qualität werden im weiteren Verlauf umfangreiche klinische Studien der Phasen 1–3 durchgeführt, in denen insbesondere die Aspekte Sicherheit, Wirksamkeit und Immunogenität untersucht werden (Abb. 3).8 Das Zulassungsverfahren von Referenzarzneimittel und Biosimilar unterscheidet sich dabei: Während das Hauptaugenmerk bei der Zulassung des Referenzarzneimittels auf dem Nachweis der klinischen Wirksamkeit und Sicherheit in der jeweils zur Zulassung angestrebten Indikation liegt, fokussiert sich die Zulassung bei Biosimilars auf den Nachweis der Äquivalenz (engl. comparability exercise). Wird diese belegt, ist von einer vergleichbaren Wirksamkeit auszugehen. Dies ist die Rationale dafür, dass – im Vergleich zur Zulassung des Referenzarzneimittels – der Umfang an klinischen Studien reduziert werden kann (Abb. 3). Nichtsdestotrotz muss in randomisierten klinischen Studien die Wirksamkeit und Sicherheit nachgewiesen werden.8
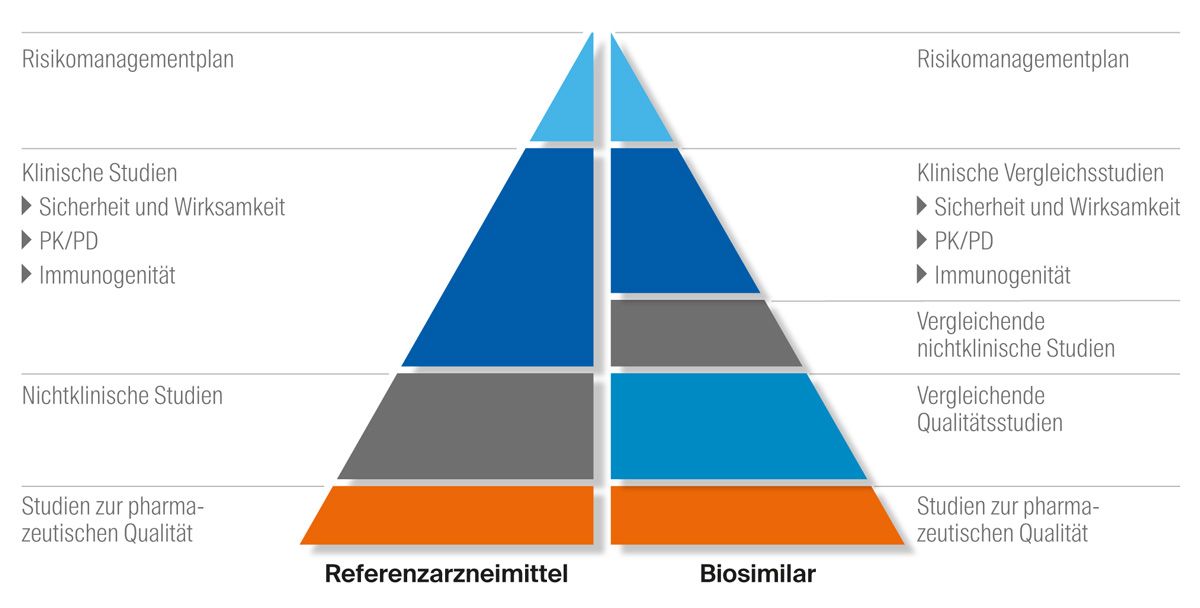
Abb. 3: Vergleich des Zulassungsprozesses von Referenzarzneimittel und Biosimilar; modifiziert nach dem Information guide for healthcare professionals der Europäischen Arzneimittel-Agentur (EMA)
Bei der Zulassung eines Biosimilars müssen der Applikationsweg, die Darreichungsform sowie die Wirkstärke mit denen des Referenzarzneimittels übereinstimmen.21 Zudem muss für die Zulassung ein umfangreiches Dossier zur Arzneimittelqualität vorgelegt werden, das die Biosimilarität belegt.21
Dossier zur Arzneimittelqualität für die Zulassung von Biosimilars (Nachweis der Biosimilarität)
- Präklinische Studien (physikochemisch, pharmakologisch-toxikologisch, Reinheitsbestimmung, biologische Aktivität)
- Klinische Studien (Pharmakokinetik/Pharmakodynamik, Wirksamkeit, Sicherheit und Immunogenität)
Exkurs: Extrapolation bei Biosimilars
Die Zulassung eines Biosimilars gilt für alle Indikationen, für die das entsprechende Referenzarzneimittel bereits zugelassen ist. Biosimilars (engl. similar = ähnlich) gehören zu den biopharmazeutischen Arzneimitteln (Biologika). Ein Biosimilar enthält eine Variante eines Wirkstoffs, mit dem in der EU bereits ein biologisches Referenzarzneimittel zugelassen ist.20,21 Aufgrund der Komplexität biologischer Arzneimittel sind Biosimilars dabei nie zu 100 % identisch mit dem Referenzarzneimittel – sie weisen aber eine sehr große Ähnlichkeit zu diesem auf (wie auch einzelne Produktchargen des Referenzarzneimittels zueinander). Da mit dem Nachweis der Äquivalenz eine vergleichbare bzw. äquivalente klinische Wirksamkeit und Sicherheit des Biosimilars belegt wird, müssen nicht für jede Indikation klinische Studiendaten vorgelegt werden. Bei der Zulassung von Biosimilars hat sich das Verfahren der Extrapolation etabliert: Dabei wird das Biosimilar in einer sensitiven Indikation im Rahmen einer klinischen Phase-3-Studie hinsichtlich der Wirksamkeit und Sicherheit untersucht und von diesen Ergebnissen und der bereits im Rahmen der zuvor durchgeführten Analyse wird auf die weiteren Indikationen basierend auf der Gesamtheit der vorhandenen Daten (engl. totality of evidence) extrapoliert, sofern sich die jeweiligen Indikationen nicht bezüglich des Wirkmechanismus (engl. mode of action) unterscheiden. Als sensitive Indikation wird eine Indikation gewählt, bei der das Auftreten von unerwünschten Wirkungen am wahrscheinlichsten ist.8 Die Extrapolation stellt ein über Jahre etabliertes, wissenschaftlich validiertes Verfahren bei der Zulassung von Biosimilars dar. Bisher ist es zu keinem Fall gekommen, in dem durch die Extrapolation Nachteile hinsichtlich der Wirksamkeit, Sicherheit und Immunogenität entstanden sind.20 Das Konzept der Extrapolation wird auch bei Änderungen im Herstellungsverfahren von Referenzarzneimitteln eingesetzt.22
Pharmakovigilanz
Auch nach der Zulassung werden Biologika intensiv hinsichtlich potenziell auftretender Nebenwirkungen und arzneimittelbezogener Probleme überwacht. Dazu gehört eine regelmäßige Neubewertung des Nutzen-Risiko-Verhältnisses. Um mögliche sicherheitsrelevante Unterschiede zwischen Biosimilar und Referenzarzneimittel zu identifizieren, die in der Zulassungsstudie nicht beobachtet wurden, sind Sicherheitsstudien nach der Zulassung (engl. post-authorisation safety studies) häufig verpflichtend. Oft muss ein Risikomanagementplan vorgelegt werden, der bei der Zulassung genehmigt wird und eine direkte und engmaschige Überwachung nach Markteinführung gewährleistet. Dies gilt auch bei der Zulassung von Biosimilars.20
Aufgrund der inhärenten Variabilität von biologischen Arzneimitteln ist besonders die Rückverfolgbarkeit auf die jeweilige Charge bedeutsam. In der EU sind die Mitgliedsstaaten dazu verpflichtet, biologische Arzneimittel bei Verdacht auf Nebenwirkungen eindeutig identifizieren zu können. Dazu müssen der Markenname, nicht nur INN(engl. international nonproprietary name)-Bezeichnung, des eingesetzten Fertigarzneimittels sowie die Chargennummer dokumentiert werden.23
4.2 Zulassungsstudie des Referenz-Natalizumab
In der zulassungsrelevanten Phase-3-Studie AFFIRM wurde die Wirksamkeit und Sicherheit des Referenz-Natalizumab untersucht.24 In der randomisierten, doppelblinden, placebokontrollierten Phase-3-Studie wurden 942 Patienten mit RRMS im Verhältnis 2:1 auf die beiden Gruppen verteilt: 627 Patienten erhielten 300 mg Natalizumab und 315 Patienten Placebo als intravenöse Infusion alle 4 Wochen über einen Zeitraum von 2 Jahren. Die Patienten waren zwischen 18 und 50 Jahren alt und hatten einen EDSS(Expanded Disability Status Scale)-Wert zwischen 0 und 5. Weiteres Einschlusskriterium war mindestens ein klinisch dokumentierter Schub im vorherigen Jahr. Als Ausschlusskriterium wurde u. a. eine Vorbehandlung mit Interferon beta oder Glatirameracetat innerhalb der letzten 6 Monate vor Studienstart definiert. Als primäre Endpunkte wurden die jährliche Schubrate nach einem Jahr sowie das Risiko einer Behinderungsprogression nach 2 Jahren eruiert.24
Natalizumab reduzierte die jährliche Schubrate nach einem Jahr signifikant um 65 % im Vergleich zu Placebo (Abb. 4). Das Risiko der Behinderungsprogression nach 2 Jahren wurde unter Natalizumab-Therapie ebenfalls signifikant um 42 % gesenkt (Abb. 4). Auch die mittels MRT nachweisbaren Läsionen nahmen durch Natalizumab im Vergleich zu Placebo signifikant ab.24
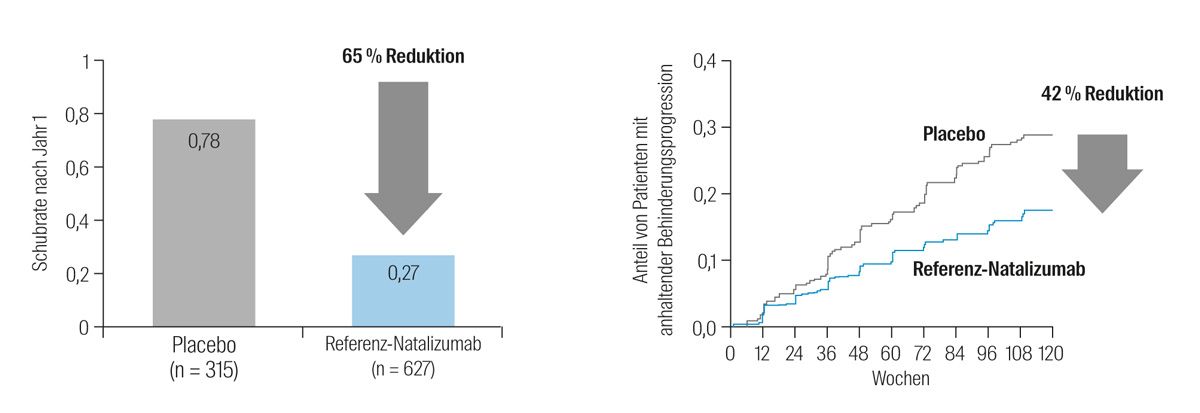
Abb. 4: Schubrate nach einem Jahr und Anteil der Patienten mit Behinderungsprogression in der Studie AFFIRM; modifiziert nach Polman et al.
Der Anteil an Patienten, der mindestens eine Nebenwirkung berichtete, war in beiden Gruppen vergleichbar (95 % bei Natalizumab vs. 96 % bei Placebo). Schwere Infektionen traten mit 3,2 % in der Natalizumab-Gruppe und mit 2,6 % in der Placebogruppe auf. Nebenwirkungen, die häufiger in der Natalizumab-Behandlungsgruppe auftraten, waren Kopfschmerzen, Fatigue und allergische Reaktionen. Überempfindlichkeitsreaktionen jedweder Art wurden bei 4 % der Patienten unter Natalizumab-Behandlung beobachtet – bei 1 % handelte es sich dabei um schwere Überempfindlichkeitsreaktionen. Zwei Patienten starben in der Natalizumab-Gruppe: ein Patient aufgrund eines Melanoms (5 Dosen Natalizumab erhalten) und ein Patient aufgrund einer Alkoholintoxikation (25 Dosen Natalizumab erhalten). 9 % der Patienten, die Natalizumab erhielten, entwickelten ADA gegen den Wirkstoff – bei 6 % wurden sie auch wiederholt detektiert. Diese Patienten zeigten verstärkte Infusionsreaktionen und eine verringerte Wirksamkeit der Therapie.24
4.3 Zulassungsstudie des Biosimilar-Natalizumab
In der zulassungsrelevanten Phase-3-Äquivalenzstudie ANTELOPE wurde das biosimilare Natalizumab hinsichtlich Wirksamkeit, Sicherheit und Immunogenität untersucht.25
In der randomisierten, doppelblinden, aktiv kontrollierten Phase-3-Studie wurden 264 Patienten mit RRMS gleichmäßig auf die beiden Therapiegruppen verteilt: 133 Patienten erhielten 300 mg des Referenz-Natalizumab (REF-NTZ) und 131 Patienten 300 mg des Biosimilar-Natalizumab (Biosim-NTZ) als intravenöse Infusion alle 4 Wochen über einen Zeitraum von 48 Wochen. Nach 24 Wochen wurde die Gruppe, die bislang das REF-NTZ erhielt, erneut randomisiert und 30 Patienten wechselten auf das Biosim-NTZ (Switch-Gruppe). Die Patienten waren zwischen 18 und 60 Jahren alt und hatten einen EDSS-Wert zwischen 0 und 5. Weiteres Einschlusskriterium war mindestens ein klinisch dokumentierter Schub im vorherigen Jahr, ≥ 1 Gadolinium-verstärkende T1-gewichtete Hirnläsion oder ≥ 9 T2-gewichtete Hirnläsionen sowie ein JCV-Index von ≤ 1,5. Der primäre Endpunkt war die kumulative Anzahl neuer aktiver Läsionen in der MRT über 24 Wochen. Sekundäre Endpunkte waren die adjustierte jährliche Schubrate, der EDSS-Wert, weitere MRT-Parameter sowie Daten zur Sicherheit und Immunogenität.25
Der modellbasierte Unterschied in der kumulativen Anzahl neuer aktiver Läsionen zwischen den beiden Gruppen in der Per-Protokoll-Population lag nach 24 Wochen bei 0,17 (0,34 Biosim-NTZ vs. 0,45 REF-NTZ) mit einem 95 %-Konfidenzintervall von −0,61 bis 0,94 (Abb. 5A). Auch die Analyse des Full Analysis Set über 48 Wochen zeigte vergleichbare Werte in der mittleren kumulativen Anzahl neuer aktiver Läsionen (Abb. 5B). Die Evaluation der sekundären Endpunkte ergab vergleichbare Ergebnisse in den beiden Gruppen: Sowohl die adjustierte Schubrate als auch der EDSS-Wert war in den 2 bzw. 3 Gruppen (einschließlich der Switch-Gruppe) auf einem ähnlichen Niveau (Tab. 2).
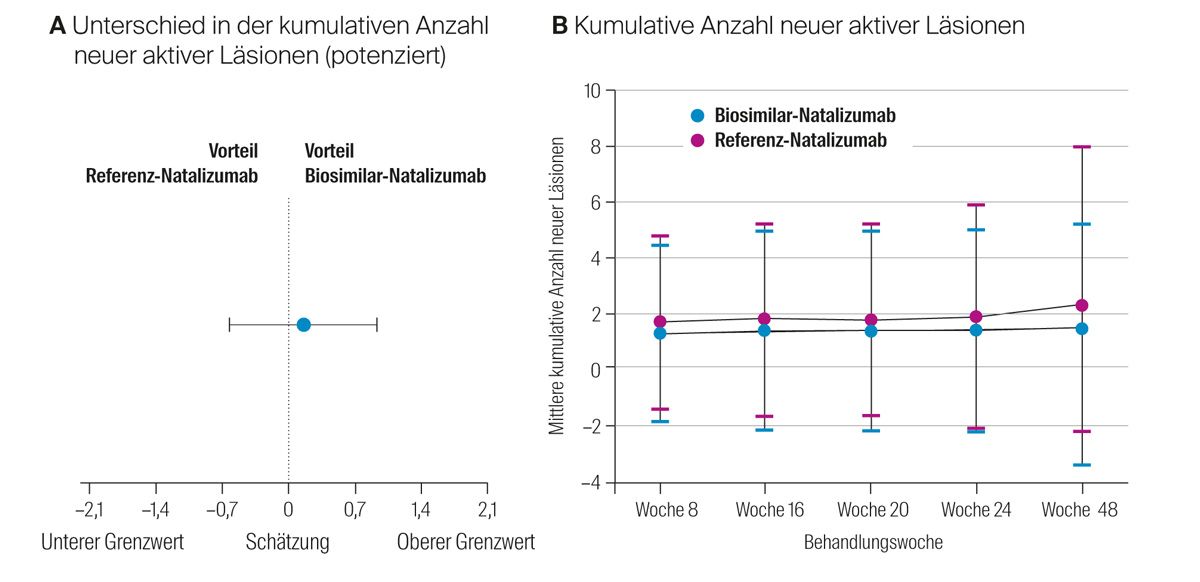
Abb. 5: Unterschied in der kumulativen Anzahl neuer aktiver Läsionen sowie kumulative Anzahl neuer aktiver Läsionen; modifiziert nach Hemmer et al.
Definition der Studienpopulation in der ANTELOPE-Studie:25
Full Analysis Set (Full Analysis Population): Umfasst alle randomisierten Studienteilnehmer, die ≥ 1 (vollständige oder anteilige) Infusion der Studienmedikation erhielten. Für die Studienauswertung wurden die Patienten gemäß der Behandlungsgruppe analysiert, in die sie randomisiert wurden.
Per-Protokoll-Population: Umfasst diejenigen Studienteilnehmer, die die 24-wöchige Behandlung ohne wesentliche Protokollabweichung abgeschlossen haben (ein Einfluss auf die Analyse des primären Endpunkts wird ausgeschlossen) und von denen ausreichende Daten aus der MRT nach Studienbeginn zur Verfügung standen.
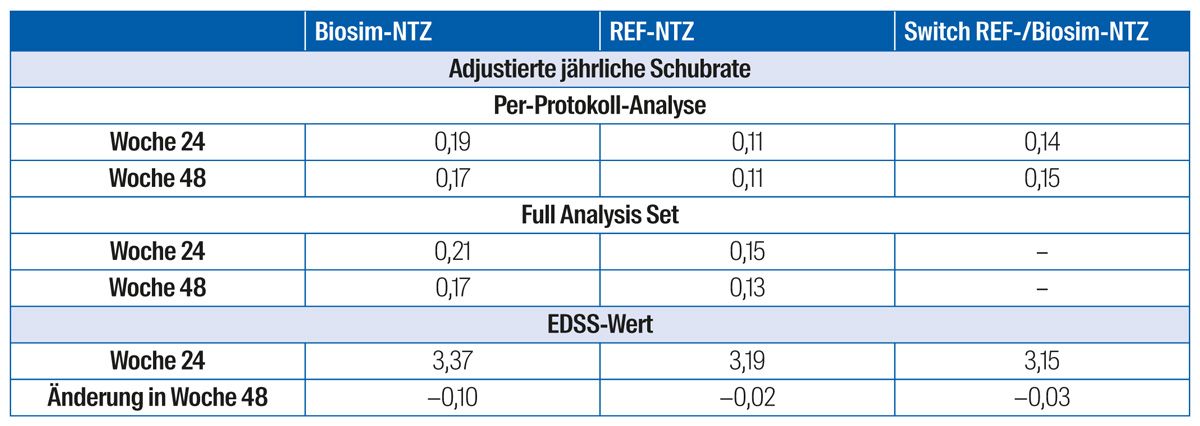
Tab. 2: Ausgewählte Daten aus der ANTELOPE-Studie; modifiziert nach Hemmer et al.
In den 3 Gruppen traten Nebenwirkungen mit einer vergleichbaren Häufigkeit auf. Die häufigsten Nebenwirkungen in allen Gruppen waren Infektionen sowie Kopfschmerzen. Es trat kein Fall von PML während der Studie und einer 24-wöchigen Nachverfolgung auf.25
Auch die Häufigkeit von ADA sowie von neutralisierenden Antikörpern (nAB) war in den beiden initialen Gruppen vergleichbar: Bei 79,4 % der Patienten, die das Biosim-NTZ erhielten, und bei 74 %, die das REF-NTZ erhielten, wurden ADA detektiert sowie nAB bei 69 % der mit dem Biosim-NTZ Behandelten und bei 66,2 % der mit REF-NTZ Behandelten. Der Wechsel vom REF-NTZ zum Biosim-NTZ war nicht mit einer gesteigerten Antikörperreaktion verbunden – Patienten, die in Woche 24 negativ für ADA oder nAB waren, blieben dies auch nach dem Wechsel.25
Die ADA-Messungen wurden mit einem hochsensitiven Elektrochemilumineszenz-Immunoassay durchgeführt, der die erforderliche hohe Sensitivität für die Immunogenitätsbewertung aufwies. Basierend auf dem Signal für den positiven Kontrollantikörper hatte der ADA-Assay eine untere Nachweisgrenze von 3,88 ng/ml für das Screening und 2,55 ng/ml für die Bestätigung.25
4.4 Vergleich der beiden Zulassungsstudien
Beim Vergleich der beiden Zulassungsstudien von Natalizumab – AFFIRM (Referenz-Biologikum) und ANTELOPE (Biosimilar) – muss auf die grundlegend verschiedene Zielsetzung hingewiesen werden. So war das Ziel der AFFIRM-Studie, die Wirksamkeit und Sicherheit von Natalizumab als neues MS-Medikament im Rahmen einer großen Phase-3-Studie zu belegen (siehe Kapitel 4.2).24 Im Vergleich dazu war die Zielsetzung der ANTELOPE-Studie eine andere: Da die Wirksamkeit von Natalizumab bereits durch die AFFIRM-Studie sowie den langjährigen Einsatz in der Praxis erwiesen war, sollte die ANTELOPE-Studie die Äquivalenz des Biosimilars gegenüber dem Referenz-Biologikum hinsichtlich Wirksamkeit, Sicherheit und Immunogenität bestätigen (siehe Kapitel 4.3). Zudem hatten bereits die Phase-1-Studiendaten eine identische Pharmakokinetik bzw. Pharmakodynamik sowie eine analytische Similarität des Biosimilars aufgezeigt, sodass von einer vergleichbaren Wirksamkeit und Sicherheit auszugehen war.26 Die unterschiedliche Zielsetzung (Äquivalenznachweis) begründet auch die kürzere Studiendauer und geringere Teilnehmerzahl in der ANTELOPE-Studie. Das Studiendesign wurde nach Abstimmung mit den Zulassungsbehörden von diesen akzeptiert und genehmigt.25
Beim direkten Vergleich von Referenz- und Biosimilar-Natalizumab wurden vergleichbare Ergebnisse hinsichtlich Wirksamkeit, Sicherheit und Immunogenität beobachtet, sodass die Bioäquivalenz des Biosimilars belegt ist (Abb. 5 und Tab. 2).25 Auf den ersten Blick verwundert die unterschiedliche Häufigkeit der detektierten ADA gegen den Wirkstoff: In der AFFIRM-Studie liegen diese im einstelligen Prozentbereich, wohingegen ADA in der ANTELOPE-Studie bei bis zu 79,4 % der Patienten gefunden wurden.24,25 Diese Diskrepanz ist auf die unterschiedliche Sensitivität der verwendeten Testsysteme zurückzuführen, das Testsystem aus dem Jahr 2023 ist wesentlich sensitiver als das aus dem Jahr 2006.16,17,24,25 Die Immunogenitätsdaten aus der ANTELOPE-Studie zeigen, dass die Häufigkeit von ADA und neutralisierenden Antikörpern in allen 3 untersuchten Gruppen – Referenz, Biosimilar und Switch – vergleichbar ist. Somit entspricht das Wirksamkeits- und Sicherheitsprofil des Biosimilar-Natalizumab dem des Referenz-Biologikums.17,25
5 Fazit
Der 2006 zugelassene Wirkstoff Natalizumab hat sich als verlaufsmodifizierende Therapie der hochaktiven RRMS bei Erwachsenen etabliert.4 Die Wirksamkeit und die Sicherheit des Wirkstoffs wurden in klinischen Studien und dem langjährigen Praxiseinsatz belegt und beruhen auf der Hemmung der transendothelialen Migration von mononukleären Leukozyten in entzündliches Parenchymgewebe.3,24,25 Auf Grundlage der Studiendaten wird Natalizumab der Wirksamkeitskategorie 3 zugerechnet.4
Durch umfassende Maßnahmen, wie die Risikostratifizierung und die regelmäßige Untersuchung/Aufklärung des Patienten, kann das PML-Risiko gut eingeschätzt und die Therapie bei PML-Verdacht frühzeitig unterbrochen werden. Weiterhin erfordert die Therapieentscheidung eine individuelle und sorgsame Nutzen-Risiko-Abwägung, insbesondere bei Schwangeren und Stillenden, bei Patienten über 65 Jahre sowie bei Kindern und Jugendlichen. Außerdem ist Natalizumab bei Patienten mit Immunschwäche, einer vorherigen PML sowie aktiven Malignomen (Ausnahme: Basaliom) kontraindiziert.3
Als Biologikum unterliegt der Wirkstoff, einschließlich seines Herstellungsprozesses, strengen Qualitätsanforderungen. Diese stellen sicher, dass die klinische Wirksamkeit und Sicherheit jeder Charge – trotz der inhärenten Mikroheterogenität der Biologika – gewährleistet sind.8,20 Die hohen Qualitätsansprüche gelten genauso für das neue Biosimilar-Natalizumab, dessen Vergleichbarkeit hinsichtlich Wirksamkeit, Sicherheit und Immunogenität in einer Phase-3-Äquivalenzstudie im Vergleich zum Referenzarzneimittel belegt wurde.23,25
Referenzen
- Faissner S et al. Progression bei multipler Sklerose – Stillstand durch fortschrittliche Behandlungen. Arzneimitteltherapie 2021; 39: 186–195
- Sorensen PS et al. The apparently milder course of multiple sclerosis: changes in the diagnostic criteria, therapy and natural history. Brain 2020; 143(9): 2637–2652
- https://www.fachinfo.de/suche/stoff/124839/Natalizumab (abgerufen am 11.01.2024)
- Hemmer B et al. Diagnose und Therapie der Multiplen Sklerose, Neuromyelitis-optica-Spektrum-Erkrankungen und MOG-IgG-assoziierten Erkrankungen, S2k-Leitlinie, 2023, in: Deutsche Gesellschaft für Neurologie (Hrsg.), Leitlinien für Diagnostik und Therapie in der Neurologie. Online: https://dgn.org/leitlinie/diagnose-und-therapie-der-multiplen-sklerose-neuromyelitis-optica-spektrum-erkrankungen-und-mog-igg-assoziierten-erkrankungen (abgerufen am 27.02.2024)
- Faissner S, Gold R. Stufentherapie der multiplen Sklerose. Medizinische Monatszeitschrift für Pharmazeuten 2023; 46(7): 230–237
- He A et al. Timing of high-efficacy therapy for multiple sclerosis: a retrospective observational cohort study. Lancet Neurol 2020; 19(4): 307–316
- Steinman L. The discovery of natalizumab, a potent therapeutic for multiple sclerosis. J Cell Biol 2012; 199(3): 413–416
- European Medicines Agency, European Commission. Biosimilars in the EU. Information guide for healthcare professionals. 2017. https://www.ema.europa.eu/en/documents/ leaflet/biosimilars-eu-information-guide-healthcare-professionals_en.pdf
- Clifford DB et al. Natalizumab-associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: lessons from 28 cases. Lancet Neurol 2010; 9(4): 438–446
- Wollebo HS et al. Persistence and pathogenesis of the neurotropic polyomavirus JC. Ann Neurol 2015; 77(4): 560–570
- Bozic C et al. Anti-JC virus (JCV) antibody prevalence in the JCV Epidemiology in MS (JEMS) trial. Eur J Neurol 2014; 21(2): 299–304
- Schippling S et al. JC virus granule cell neuronopathy and GCN-IRIS under natalizumab treatment. Ann Neurol 2013; 74(4): 622–626
- Agnihotri SP et al. JCV GCN in a natalizumab-treated MS patient is associated with mutations of the VP1 capsid gene. Neurology 2014; 83(8): 727–732
- Dong-Si T et al. Outcome and survival of asymptomatic PML in natalizumab-treated MS patients. Ann Clin Transl Neurol 2014; 1(10): 755–764
- Ho PR et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: a retrospective analysis of data from four clinical studies. Lancet Neurol 2017; 16(11): 925–933
- https://www.pei.de/SharedDocs/arzneimittel/sera/EU-1-06-346-001.html (abgerufen am 14.03.2024)
- https://www.pei.de/SharedDocs/arzneimittel/sera/EU-1-23-1745.html (abgerufen am 14.03.2024)
- Scarpazza C et al. Should frequent MRI monitoring be performed in natalizumab-treated MS patients? A contribution to a recent debate. Mult Scler 2020; 26(10): 1227–1236
- Wattjes MP et al. MAGNIMS study group. Evidence-based guidelines: MAGNIMS consensus guidelines on the use of MRI in multiple sclerosis – establishing disease prognosis and monitoring patients. Nat Rev Neurol 2015; 11(10): 597–606
- Leitfaden der Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ): Biosimilars. 2. Auflage, Version 1.0; Januar 2021
- European Medicines Agency (EMA), European Commission: Guideline on similar biological medicinal products. 2014. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-rev1_en.pdf
- Weise M E et al. Biosimilars: the science of extrapolation. Blood 2014; 124: 3191–3196
- European Medicines Agency (EMA): Committee for Medicinal Products for Human Use (CHMP): Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/01/WC500180219. pdf
- Polman CH et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med 2006; 354(9): 899–910
- Hemmer B et al. Efficacy and Safety of Proposed Biosimilar Natalizumab (PB006) in Patients With Relapsing-Remitting Multiple Sclerosis: The Antelope Phase 3 Randomized Clinical Trial. JAMA Neurol 2023; 80(3): 298–307
- Wessels H et al. Pharmacokinetic and pharmacodynamic similarity of biosimilar natalizumab (PB006) to its reference medicine: a randomized controlled trial. Expert Opin Biol Ther. 2023 Jul-Dec; 23(12): 1287–1297.
Bildquellen
Titel: © William – stock.adobe.com
Tutorielle Unterstützung
Die tutorielle Unterstützung der Fortbildungsteilnehmer erfolgt durch unseren ärztlichen Leiter Dr. med. Alexander Voigt in Zusammenarbeit mit der arztCME-Redaktion. Inhaltliche Fragen können über das Kommentarfeld, direkt per Mail an service@arztcme.de oder via Telefon unter Tel.: +49(0)180-3000759 gestellt werden. Inhaltliche Fragen werden von unserem ärztlichen Leiter bzw. nach Rücksprache mit diesem und evtl. dem Autor auch von der arztCME-Redaktion beantwortet.
Technischer Support
Der technische Support der Online-Akademie arztCME.de erfolgt durch geschulte Mitarbeiterinnen und Mitarbeiter des Betreibers health&media GmbH unter der E-Mail-Adresse technik@arztcme.de oder via Telefon unter Tel.: 49(0)180-3000759.