Multidisziplinäre Versorgung bei der Spinalen Muskelatrophie: Fokus auf der ambulanten Versorgung in der Praxis und im neuromuskulären Zentrum
Interessengebiete: Allgemeinmedizin und Innere Medizin, Kinder- und Jugendmedizin, Neurologie
Spinale Muskelatrophien sind seltene, genetisch bedingte Erkrankungen des motorischen Nervensystems, die zu fortschreitender Muskelschwäche führen. Je nach Typ beginnt die Erkrankung bereits im Säuglingsalter und kann unbehandelt lebensbedrohlich verlaufen. Neue Therapieansätze bieten jedoch Hoffnung auf eine langfristige Verbesserung der motorischen Funktionen und der Lebensqualität der Betroffenen. Diese CME-Fortbildung sensibilisiert für die Früherkennung, informiert über Behandlungsmöglichkeiten und betont die Bedeutung der Überweisung an spezialisierte Zentren.
Kursinhalt
Inhaltsverzeichnis
Vorwort
Die 5q-assoziierte spinale Muskelatrophie (5q-SMA) ist eine seltene, autosomal-rezessiv vererbte neurodegenerative Erkrankung. Sie führt durch den fortschreitenden Verlust von Alpha-Motoneuronen im Rückenmark zu Lähmungen, die vor allem die proximalen Extremitäten und die Rumpfmuskulatur betreffen, während die kognitive Funktion dabei unbeeinträchtigt bleibt. Die genaue Ausprägung der Symptome und die Lebenserwartung variieren abhängig vom jeweiligen SMA-Typ, wobei die schwerste Form bereits im Säuglingsalter auftritt und unbehandelt in den ersten 6 Monaten zum Tod führt. Neue Therapieansätze bieten jedoch Hoffnung auf eine langfristige Verbesserung der motorischen Funktionen und damit auf eine Verbesserung des allgemeinen Gesundheitszustandes und der Lebensqualität.
Eine frühzeitige Diagnose, ein kontinuierliches Monitoring und der Zugang zu geeigneten Therapien sind entscheidend für die Versorgung von Patient*innen mit 5q-SMA. Daher ist es wichtig, über die Erkrankung, ihre Symptome und Behandlungsmöglichkeiten aufzuklären.
Diese CME-Fortbildung richtet sich an Ärzt*innen unterschiedlicher Fachrichtungen mit dem Ziel, die Ärzt*innen für die 5q-SMA als mögliche Ursache verschiedener Symptome zu sensibilisieren, über Behandlungsmöglichkeiten zu informieren und die Relevanz der Überweisung an ein neuromuskuläres Zentrum aufzuzeigen.
Hintergrund
Die 5q-assoziierte spinale Muskelatrophie (5q-SMA) ist eine seltene neurodegenerative Erkrankung, die durch fortschreitenden Muskelschwund und Lähmungen gekennzeichnet ist. Sie zählt zu den häufigsten autosomal-rezessiv vererbten Erkrankungen [1,2]. In Deutschland liegt die Inzidenz bei etwa 1:7.000, was einer geschätzten Anzahl von rund 1.500 Betroffenen entspricht [3]. Etwa eine von 40 Personen in der Allgemeinbevölkerung ist heterozygoter Träger des genetischen Risikos, was mit einem erhöhten Erkrankungsrisiko bei Geschwistern von Patient*innen mit 5q-SMA assoziiert ist [4].
Der Erkrankung liegt ein biallelischer Gendefekt zugrunde, der zu einem Mangel an Survival-of-Motor-Neuron (SMN)-Protein führt [5,6]. Dieser Mangel führt pathophysiologisch zu einer reduzierten Integrität von Cytoskelett, Synapsen und axonalem Transport in spinalen Alpha-Motoneuronen, was schließlich zur axonalen Degeneration und Absterben der Neurone führt. Dies wiederum bedingt eine Atrophie der von den Neuronen versorgten Muskeln mit folgenden Paresen.
Die daraus resultierende Muskelschwäche führt im weiteren Verlauf der Erkrankung zu einer zunehmenden Beeinträchtigung der motorischen, respiratorischen und bulbären Funktionen [7] unterscheiden sich Erkrankungsbeginn, Schweregrad der Symptomatik und Krankheitsverlauf abhängig vom SMA-Typ – in den meisten Fällen ist die Lebensqualität stark beeinträchtigt und die Lebenserwartung reduziert [4]. In den letzten Jahren haben neue therapeutische Ansätze die Behandlungsmöglichkeiten wesentlich erweitert und die Versorgungssituation der Betroffenen grundlegend verändert. Zu den krankheitsmodifizierenden derzeit zugelassenen Therapieoptionen zählen das Antisense-Oligonukleotid Nusinersen [8] sowie der Splicing-Modulator Risdiplam [9] und das gentherapeutische Arzneimittel Onasemnogen Abeparvovec [10]. Diese medikamentösen Innovationen ermöglichen nicht nur eine Stabilisierung, sondern potenziell auch eine Verbesserung der motorischen Funktion bis hin zu einer weitgehend altersentsprechenden Entwicklung in einigen Fällen, was sich positiv auf Lebensqualität und Prognose auswirkt [11].
Genetische Ursache
Als genetische Ursache der 5q-SMA wurde eine Defektmutation oder Deletion des SMN1-Gens identifiziert. Das SMN1– und das SMN2-Gen sind zwei benachbarte Gene auf dem q-Arm von Chromosom 5, die für das SMN-Protein kodieren. Bei gesunden Personen wird das SMN-Protein primär durch das SMN1-Gen exprimiert, das für etwa 90 % der Gesamtmenge an SMN-Protein verantwortlich ist. Dabei entsteht zunächst eine SMN1-prä-mRNA, die korrekt gespleißt wird und das Exon 7 enthält – eine essenzielle Voraussetzung für die Produktion eines voll funktionsfähigen SMN-Proteins. Folglich ist 100 % des über das SMN1-Gen exprimierten SMN-Proteins funktionsfähig (Abb. 1) [5].
Das SMN2-Gen ist nahezu identisch mit dem SMN1-Gen und tritt in variabler Kopienzahl auf. Aufgrund eines Einzelnukleotid-Polymorphismus im Exon 7 kommt es zu einem alternativen Spleißen und dadurch zu einem Verlust von Exon 7 bei dem Großteil der SMN2-mRNA [5]. Infolgedessen trägt SMN2 nur zu etwa 10 % zur Produktion funktionaler SMN-Proteine bei, während der Großteil der translatierten Proteine instabil und nicht funktionsfähig ist [6].
Bei 5q-SMA führt die Mutation im SMN1-Gen zu einem Mangel an SMN-Protein und folglich zur Neurodegeneration von Alpha-Neuronen im Rückenmark und Hirnstamm. Dieser Mangel kann nur teilweise durch die Expression von SMN2 kompensiert werden. Die Anzahl der SMN2-Kopien ist dabei entscheidend für den Schweregrad der 5q-SMA: Je weniger Kopien vorhanden sind, desto schwerer ist in der Regel der Verlauf der 5q-SMA [5,11].
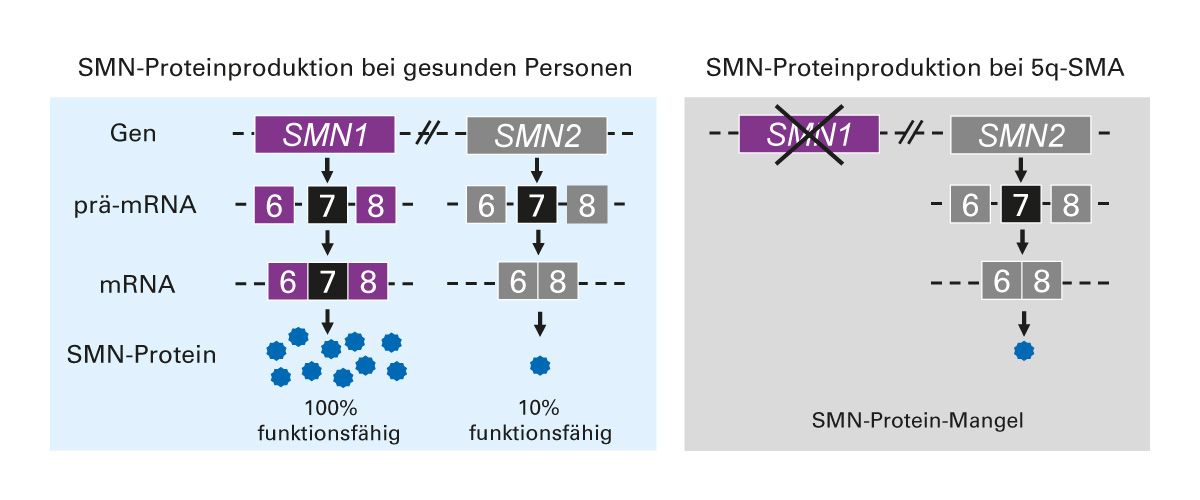
Abb. 1: SMN-Proteinproduktion bei gesunden Personen (links) und Personen mit 5q-SMA (rechts) [5,6]. Abbildung modifiziert nach d’Ydewalle et al.[5] 5q-SMA: 5q-assoziierte spinale Muskelatrophie; SMN: Survival-of-Motor-Neuron
Klassifizierung
Die Einteilung der 5q-SMA in fünf Typen (Typ 0-4) basierte zunächst auf dem Erkrankungsbeginn, der Lebenserwartung und den voraussichtlich zu erreichenden motorischen Meilensteinen. Typ 0 stellt die schwerste Verlaufsform dar, mit ersten Symptomen bereits intrauterin und einer Lebenserwartung von meist weniger als sechs Monaten. Von Typ 0 bis zu Typ 4 steigen sowohl das Alter bei Symptombeginn als auch die Lebenserwartung, sodass Typ 4 häufig erst im dritten Lebensjahrzehnt auftritt [11].
Durch den Einsatz krankheitsmodifizierender Therapien kann der Krankheitsverlauf jedoch vom ursprünglich definierten SMA-Typ abweichen, insbesondere in Abhängigkeit vom Zeitpunkt des Therapiebeginns. Daher wird nun zusätzlich der funktionelle Status bei Diagnosestellung berücksichtigt und zwischen Non-Sitter, Sitter und Walker unterscheiden. Diese Einteilung erfasst jedoch präsymptomatische Patient*innen nicht angemessen, da sie zum Zeitpunkt der Diagnose noch keine klinischen Symptome zeigen [11,12].
Die in den aktuellen Leitlinien dokumentierte, neu entwickelte, mehrdimensionale Klassifikation integriert daher zusätzlich die Anzahl der SMN2-Kopien, da diese eine prognostische Aussage über den zu erwartenden Phänotyp präsymptomatischer Patient*innen ermöglicht. Somit berücksichtigt dieses Klassifikationsmodell nicht nur den SMA-Typ (Typ 0-4) und den aktuellen motorischen Funktionsstatus (Non-Sitter, Sitter, Walker), sondern auch die genetischen Faktoren zur genaueren Verlaufseinschätzung (SMN2-Kopienzahl) [11].
Symptomatik der SMA
Symptomatisch äußert sich die SMA typischerweise durch generalisierte Paresen, die vor allem die rumpfnahen Muskeln und die Beine betreffen, während die kognitive Funktion unbeeinträchtigt bleibt. Die Symptome variieren jedoch erheblich je nach SMA-Typ, vom Typ 0 mit schweren Symptomen bereits bei Geburt und letalem Verlauf bis zu Typ 4 mit Symptomfreiheit meistens bis zum dritten Lebensjahrzehnt und einem langsamen Verlauf (Abb. 2) [11,12].
Respiratorische Komplikationen und ein erhöhtes Erstickungsrisiko sind bei SMA-Patient*innen eine der Hauptursachen für Krankenhausaufenthalte, Morbidität und Mortalität [13,14]. Neben der Atemfunktion kann auch die Kau- und Schluckfunktion beeinträchtigt sein, was wiederum zu unzureichender Nahrungsaufnahme und Wachstumsstörungen führt [13]. Zusätzlich treten häufig orthopädische Begleiterkrankungen wie Gelenkkontrakturen und Skoliosen auf, insbesondere bei fortschreitender Erkrankung [15,16].
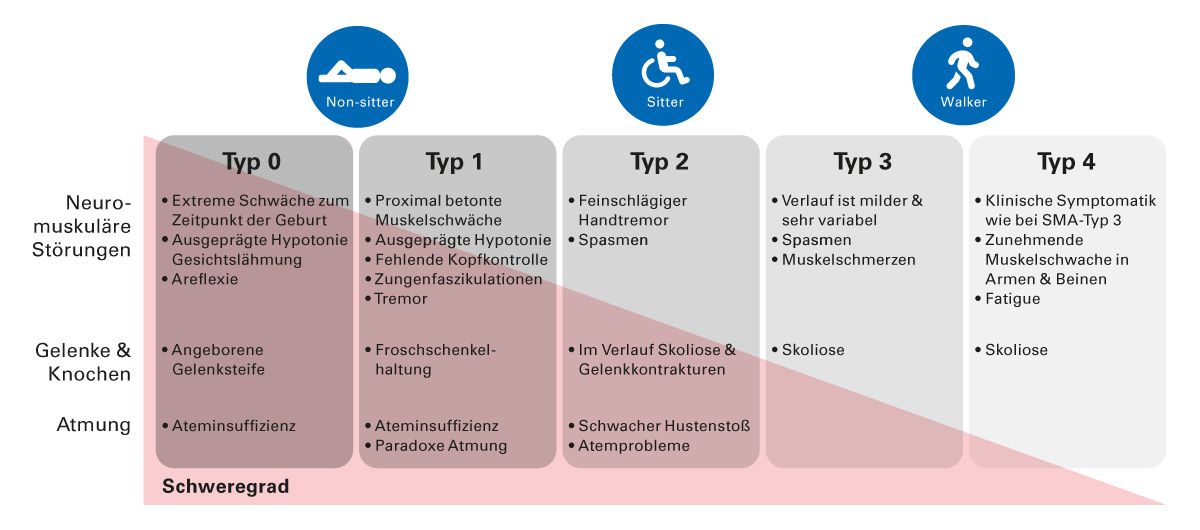
Abb. 2: Unterschiedliche Symptomatik abhängig vom SMA-Typ [11,13]
Diagnose
Eine frühe Diagnose und ein rascher Therapiebeginn sind entscheidend für den Therapieerfolg. Die Diagnosestellung erfolgt durch Anamnese, klinische Untersuchung und molekulargenetische Analyse der SMN1– und SMN2-Kopien [11].
Die klinischen Symptome einer 5q-SMA variieren je nach Altersgruppe in Art und Schweregrad. Bei Säuglingen sind muskuläre Hypotonie („Floppy Infant“), mangelnde Kopfkontrolle, Zungenfaszikulationen, paradoxe Atmung und Froschschenkelhaltung charakteristisch [11,14]. Bei älteren Kindern treten häufig Dysphagie, Kauprobleme, fehlende tiefe Sehnenreflexe, Spasmen und Skoliose auf. Das Nichterlernen oder der Verlust motorischer Meilensteine ist ein weiteres zentrales Warnzeichen [17]. Jugendliche und Erwachsene zeigen meist progrediente Muskelschwäche in Armen und Beinen. Zudem sollten Fatigue und Kurzatmigkeit beachtet werden [13].
Bei klinischem Verdacht auf eine 5q-SMA muss umgehend eine molekulargenetische Diagnostik veranlasst werden. Die Bestimmung der SMN1– und SMN2-Kopienzahl (Exons 7 und 8) sollte mittels quantitativer Verfahren erfolgen, wobei die Multiplex Ligation-dependent Probe Amplification (MLPA) aktuell als Goldstandard gilt. Die Anzahl der SMN1-Kopien ist für die Diagnose entscheidend: Eine homozygote Deletion der Exons 7 und/oder 8 bestätigt eine 5q-SMA. Das Fehlen einer homozygoten Deletion schließt eine 5q-SMA jedoch nicht aus. In diesem Fall sollte eine Sequenzierung des SMN1-Gens (Sanger-Sequenzierung oder Long-Read-NGS) durchgeführt werden, um heterozygote pathogene Varianten zu identifizieren. Falls keine solche Mutation vorliegt, sollte eine andere Form der SMA oder neuromuskulären Erkrankung (NMD) mittels NMD-Genpanel oder Vollständiger Exom- oder Gensequenzierung (WES/WGS) abgeklärt werden (Abb. 3) [11].
Nach gesicherter Diagnose einer 5q-SMA ist die Bestimmung der SMN2-Kopienzahl essenziell, da sie Rückschlüsse auf den Schweregrad der Erkrankung zulässt und eine wichtige Entscheidungsgrundlage für die Therapie darstellt [11].
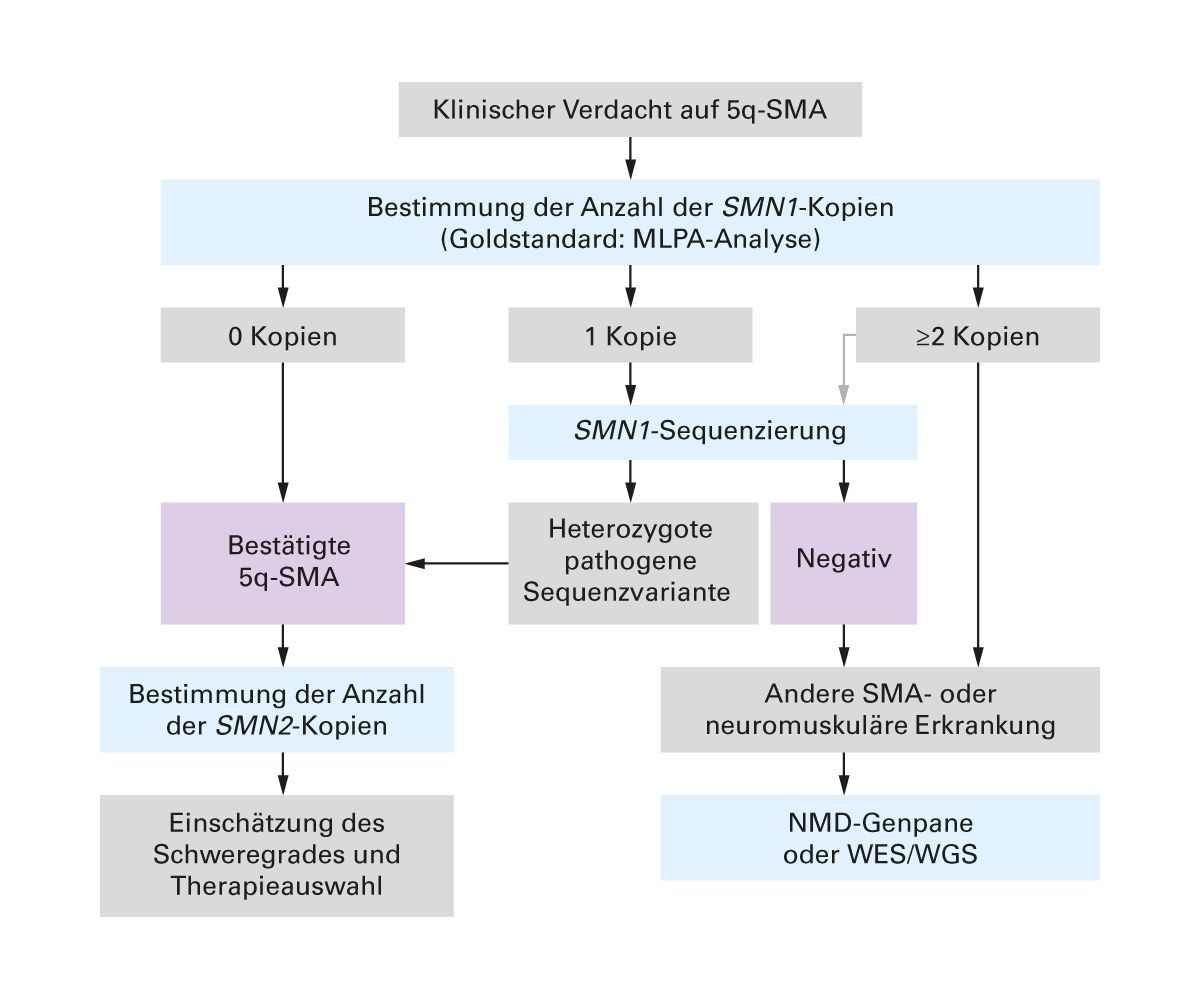
Abb. 3: Molekulargenetische Testung zur Bestätigung der 5q-SMA-Diagnose [11]. Abbildung modifiziert nach S2k-Leitlinien zur Diagnostik und Therapie der 5q-assoziierten spinalen Muskelatrophie im Kindes- und Erwachsenenalter (Stand 31.12.2024)[11] 5q-SMA: 5q-assoziierte spinale Muskelatrophie; MLPA-Analyse: Multiplex Ligation-dependent Probe Amplification-Analyse; NMD-Genpanel: Genpanel für neuromuskuläre Erkrankungen; SMN: Survival-of-Motor-Neuron; WES: Vollständige Exomsequenzierung; WGS: Vollständige Genomsequenzierung
Eine wesentliche Neuerung in der SMA-Diagnostik ist die Aufnahme der 5q-SMA in das allgemeine Neugeborenen-Screening in Deutschland, die im Oktober 2021 vom Gemeinsamen Bundesausschuss beschlossen wurde [11]. Das Screening auf eine homozygote Deletion im Exon 7 des SMN1-Gens erfolgt nach Einwilligung der Eltern im Rahmen des allgemeinen Neugeborenen-Screenings und wird mit der gleichen Filterkarte wie die anderen Screeninguntersuchungen durchgeführt. Da jedoch nicht alle Eltern dem Screening zustimmen, oder bei Geburten außerhalb Deutschlands, wird bei klinischem Verdacht dringend eine Testung empfohlen [11]. Nach einem positiven Befund im Neugeborenen-Screening sollte für die Diagnosestellung umgehend eine Überweisung an ein spezialisiertes neuromuskuläres Zentrum erfolgen [11].
Therapieoptionen
In Deutschland sind derzeit drei medikamentöse Therapien zur Behandlung der 5q-SMA zugelassen: Nusinersen, Risdiplam und Onasemnogene Abeparvovec. Diese kausal orientierten Therapien ermöglichen die kontinuierliche Produktion des SMN-Proteins in den Motoneuronen des Rückenmarks, die unbehandelt degenerieren würden [18]. Dabei wird zwischen reversiblen Therapien, die die Genexpression auf mRNA-Ebene beeinflussen, und der Genersatztherapie unterschieden, die dauerhaft in die Genexpression eingreift [11].
Reversible Therapieansätze wie Nusinersen und Risdiplam setzen an der Modulation des Spleißvorgangs des SMN2-Gens an, um die Inklusion von Exon 7 zu fördern und die Produktion funktionaler SMN-Proteine zu erhöhen (Abb. 4) [6,12].
Nusinersen ist ein Antisense Oligonukleotide (ASO) und wurde als erste krankheitsmodifizierende Therapie für die 5q-SMA zugelassen. Die Zulassung umfasst alle Altersgruppen, Erkrankungsstadien und Schweregrade [8]. Klinische Studien belegten eine Verbesserung der motorischen Funktion bei Säuglingen und Kindern [19,20], wobei der Therapieerfolg maßgeblich vom Behandlungszeitpunkt und vom Stadium der Erkrankung abhing [11]. Auch bei Erwachsenen liegen inzwischen vermehrt Real-World-Erfahrungen zur Therapie mit Nusinersen vor, die darauf hinweisen, dass eine gute Wirksamkeit besteht [21]. Aufgrund der wiederholten intrathekalen Injektion, die nach anfänglicher Aufsättigung alle 4 Monate erfolgt, wird die Therapie in neuromuskulären Zentren mit Erfahrung in der Behandlung von SMA empfohlen [8,11].
Risdiplam, ein täglich oral verabreichtes Small Molecule, steigert ebenfalls die Produktion von SMN-Proteinen [9,11]. Aufgrund des in klinischen Studien beobachteten bedeutsamen klinischen Nutzen bei infantiler 5q-SMA, wurde die Zulassung 2023 für die Anwendung direkt nach der Geburt erweitert [11,22]. Obwohl Risdiplam für alle SMA-Patient*innen zugelassen ist, zeigten Studien bei later-onset SMA eine womöglich etwas weniger ausgeprägte Verbesserung der motorischen Funktion. Direkte Vergleichsstudien mit Nusinersen liegen bislang nicht vor, und in die bisherigen Untersuchungen wurden nur sehr wenige ältere und tendenziell auch schwerer betroffene SMA-Patient*innen eingeschlossen. Daher ist eine direkte Aussage zum Vergleich der Wirksamkeit von Risdiplam und Nusinersen nicht möglich [23]. Auch hier wird die Durchführung in neuromuskulären Zentren mit Expertise in der Behandlung der 5q-SMA empfohlen [11].
Die Genersatztherapie Onasemnogen Abeparvovec nutzt einen Adeno-assoziierten Virus-Vektor, der eine funktionsfähige Kopie des humanen SMN1-Gens enthält [10,11]. Nach intravenöser Gabe gelangt der Vektor über die noch unreife Blut-Hirn-Schranke in den ersten sechs Lebensmonaten ins zentrale Nervensystem (ZNS). Durch diesen irreversiblen Eingriff wird die SMN1-cDNA als Episom in die Alpha-Motoneurone eingeschleust und ermöglicht eine kontinuierliche SMN-Proteinproduktion [11]. Seit 2020 besteht eine Zulassung in Europa für Patient*innen mit einer gesicherten SMN1-Genmutation und klinischer Diagnose eines SMA-Typ 1 (unabhängig von der SMN2-Kopienzahl) oder SMA mit ≤3 SMN2-Kopien (unabhängig vom SMA-Typ) [10,11]. Auch hier wurde in klinischen Studien eine Verbesserung der motorischen Funktion bei Säuglingen und Kindern mit 5q-SMA beobachtet [24]. Die intravenöse Verabreichung muss in spezialisierten Zentren unter strengen Sicherheitsvorgaben erfolgen. In der Fachinformation zu Onasemnogen Abeparvovec wird darauf hingewiesen, dass derzeit nur begrenzte Erfahrungen zur Wirksamkeit und Sicherheit bei Kindern über 2 Jahren und mit einem Körpergewicht über 13,5 kg vorliegen [10]. Studien legen nahe, dass mit zunehmendem Gewicht das Risiko für Nebenwirkungen steigen kann, möglicherweise bedingt durch die höhere Dosierung sowie das weiterentwickelte Immunsystem [25]. Daher ist es empfehlenswert, die Therapie frühzeitig zu beginnen [11]. Der aktuelle europäische Konsensusbericht spricht sich zudem gegen den Einsatz von Onasemnogen Abeparvovec bei Erwachsenen aus [11,26].
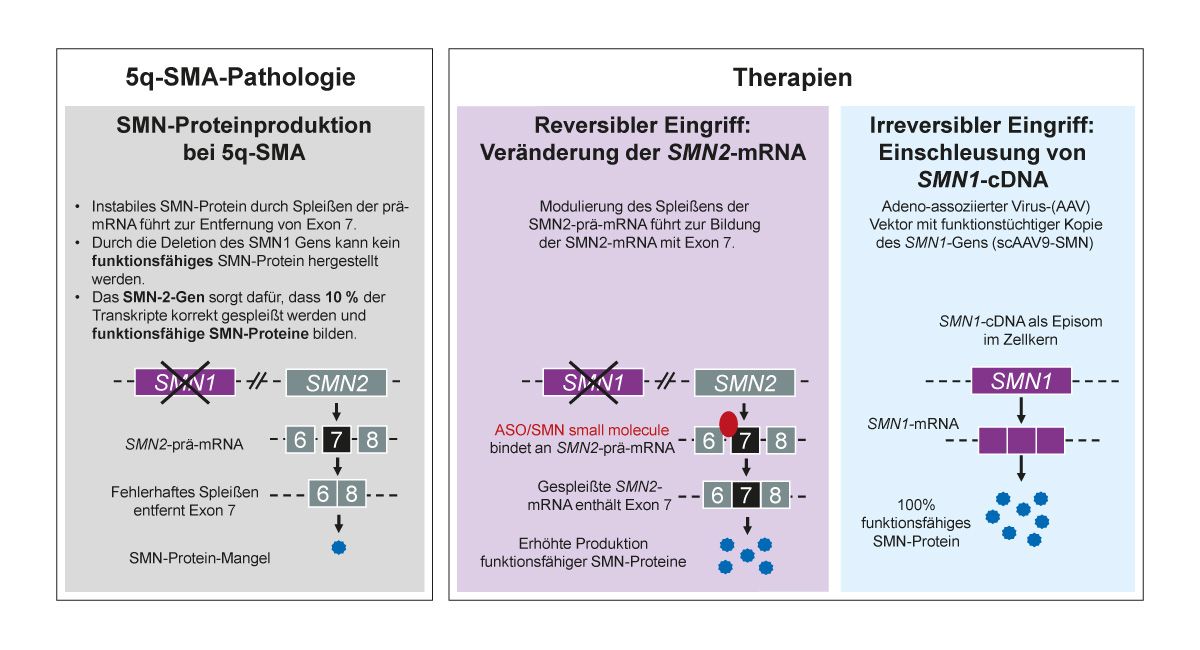
Abb. 4: Wirkmechanismen der medikamentösen Therapien [5,6,8-10,12]. Abbildung modifiziert nach Day et al. [12] 5q-SMA: 5q-assoziierte spinale Muskelatrophie; AAV: Adeno-assoziierter Virus; mRNA: Messenger RNA; scAAV9-SMN: AAV-Vektor mit SMN1-Genkopie; SMN: Survival-of-Motor-Neuron. ASO: Antisense-Olegonukleotid
Familienplanung
Dank dieser neuen Therapieoptionen kann nicht nur die Progression der 5q-SMA maßgeblich verlangsamt werden, sondern immer mehr Menschen mit 5q-SMA erreichen ein fortpflanzungsfähiges Alter. Daher gewinnt die Familienplanung von Patient*innen mit 5q-SMA zunehmend an Bedeutung und stellt einen relevanten Faktor in der therapeutischen Entscheidungsfindung dar [11]. Zu den krankheitsmodifizierenden Therapien für erwachsene Patient*innen zählen insbesondere Risdiplam und Nusinersen [11]. Da tierexperimentelle Studien zu Risdiplam auf ein mögliches Risiko für die Fruchtbarkeit bei Männern sowie auf eine potenzielle embryo-fetale Toxizität bei Frauen hindeuten, ist eine zuverlässige Verhütung bei Patient*innen, die Risdiplam einnehmen, zwingend erforderlich. Im Falle eines Kinderwunschs wird daher empfohlen, die Behandlung mit Risdiplam bei Männern mindestens vier Monate, bei Frauen mindestens einen Monat vor der Zeugung zu unterbrechen. Eine Kryokonservierung von Spermien sollte vor Therapiebeginn mit Risdiplam bei erwachsenen Patienten erwogen werden. Auch während der Stillzeit sollte Risdiplam nicht angewendet werden, da in Tierstudien eine Ausscheidung des Wirkstoffs und seiner Metaboliten in die Muttermilch nachgewiesen wurde [9,11]. Für Nusinersen konnten in tierexperimentellen Studien bislang weder Auswirkungen auf die Fruchtbarkeit noch embryo-fetale Toxizität festgestellt werden. Allerdings liegen bisher nur sehr begrenzte Erfahrungen zur Anwendung während der Schwangerschaft vor [8]. Aus Vorsichtsgründen sollte daher auch Nusinersen während Schwangerschaft und Stillzeit möglichst nicht eingesetzt werden [8,11].
Beurteilung und Überwachung motorischer Funktionen
Um den Therapieerfolg und die allgemeine motorische Entwicklung bei 5q-SMA zu überwachen, sollten mindestens einmal jährlich, unter medikamentöser Therapie sogar alle vier bis sechs Monate, die motorischen Funktionen von geschultem Fachpersonal untersucht werden [11].
Vor der Diagnose helfen diese Untersuchungen, Kinder zu identifizieren, die wichtige motorische Meilensteine nicht erreichen [27]. Nach der Diagnose liefern sie Informationen über den SMA-Typ und den Schweregrad der Erkrankung [28]. Zudem ermöglichen sie die Überwachung des Krankheitsverlaufs während der Therapie und somit die Beurteilung des Behandlungserfolgs [29]. Außerdem helfen sie, den richtigen Zeitpunkt für notwendige therapeutische Maßnahmen, wie Physiotherapie oder den Einsatz von Hilfsmitteln zu bestimmen [28].
Die Untersuchungen sollen Muskelkraft, Gelenkbeweglichkeit und funktionelle Einschränkungen im Alltag erfassen. Welche Tests angewendet werden, hängt vom Alter, dem motorischen Funktionsniveau und dem Krankheitsverlauf ab [11]. Bei Non-Sittern werden die CHOP INTEND-Skala (Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders) für Kinder und die CHOP ATEND-Skala (Children’s Hospital of Philadelphia Adult Test of Neuromuscular Disorders) für Erwachsene genutzt, um grundlegende motorische Fähigkeiten zu messen [11,30]. Bei Sittern sind die Beweglichkeit und Funktion der oberen Extremitäten besonders wichtig, weshalb Tests wie RULM (Revised Upper Limb Module) und HFMSE (Hammersmith Functional Motor Scale Expanded) angewendet werden [11,31,32]. Bei Walkern hingegen sind die oberen Extremitäten meist weniger stark betroffen, sodass ihre Gehfähigkeit im Fokus steht. Neben RULM und HFMSE werden hier Tests wie der 6-Minuten-Gehtest oder der Timed-Up-and-Go-Test, bei starken Einschränkungen auch der 10-Meter-Gehtest eingesetzt [11,33].
Allerdings wurden die verwendeten Funktionsscores für unbehandelte 5q-SMA entwickelt und ermöglichen eine gute Unterscheidung sehr schwacher Patient*innen. Körperlich fittere SMA-Patient*innen erreichen trotz deutlicher motorischer Defizite oft die maximale Punktzahl. Durch diesen möglichen „Ceiling“-Effekt können Therapieeffekte nicht immer genau erfasst werden [11].
Datendokumentation in Registern
Ein zentrales Instrument der Versorgungsforschung im Bereich der 5q-SMA ist das SMArtCARE-Register. Dieses krankheitsspezifische Register dokumentiert systematisch den Krankheitsverlauf von Patient*innen mit 5q-SMA, die mit Nusinersen, Onasemnogen Abeparvovec oder Risdiplam behandelt werden. Ziel ist es, umfassende Real-World-Daten zur Langzeitwirksamkeit und -sicherheit dieser neuen Therapien zu gewinnen [11,34]. Die so gewonnenen Erkenntnisse tragen maßgeblich zur evidenzbasierten Weiterentwicklung der Versorgung bei und schließen eine wichtige Lücke zwischen klinischer Forschung und praktischer Anwendung im Versorgungsalltag [34].
Insbesondere bei neuartigen, hochspezifischen Therapien ist eine anwendungsbegleitende Datenerhebung nicht nur wissenschaftlich bedeutsam, sondern auch regulatorisch erforderlich. In bestimmten Fällen stellt der Gemeinsame Bundesausschuss die Teilnahme an einer Datenerhebung als Auflagen oder Vereinbarungen für den therapeutischen Einsatz fest. So ist die Dokumentation der Therapieergebnisse im SMArtCARE Register [35] für Onasemnogen Abeparvovec seit Februar 2022 [36] und für Risdiplam seit Oktober 2024 [37] verpflichtend festgelegt. Für Nusinersen hingegen ist die Teilnahme am Register freiwillig. Dennoch stellt die Dokumentation auch hier eine wichtige Grundlage für die kontinuierliche Bewertung von Nutzen und Risiken der Therapie dar. Die freiwillige Beteiligung von Behandelnden und Patient*innen am SMArtCARE-Register trägt wesentlich zur Transparenz und Qualitätssicherung in der Versorgung bei.
Multidisziplinäre Versorgung in neuromuskulären Zentren
Die 5q-SMA ist eine komplexe Erkrankung, die eine umfassende, multidisziplinäre Versorgung erfordert. Eine Schlüsselrolle übernehmen dabei spezialisierte neuromuskuläre Zentren. In diesen Einrichtungen stellen erfahrene Fachärzt*innen eine rasche Diagnose sicher, führen regelmäßige klinische Untersuchungen durch und entwickeln individuell angepasste Therapiepläne mit kontinuierlichem Monitoring [38,39].
Darüber hinaus profitieren Betroffene vom Zugang zu innovativen Therapien und klinischen Studien.
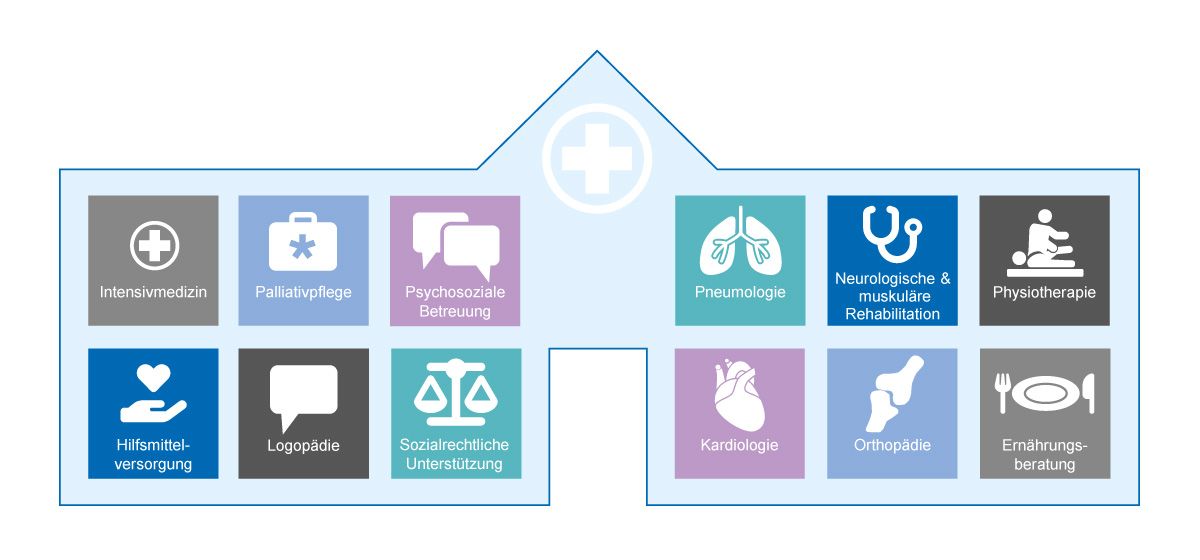
Ein zentrales Merkmal der neuromuskulären Zentren ist die enge Zusammenarbeit verschiedener Fachdisziplinen – ein entscheidender Faktor für die erfolgreiche Behandlung einer so vielschichtigen Erkrankung wie der 5q-SMA (Abb. 5). Studien belegen, dass eine spezialisierte multidisziplinäre Betreuung sowohl die motorischen Fähigkeiten als auch die Lebensqualität von SMA-Patient*innen deutlich verbessern kann [38].
Bildquelle:
Pneumologie
Eine sorgfältige und regelmäßige Überwachung der Lungenfunktion ist aufgrund der häufig auftretenden Atemprobleme besonders wichtig [11].
Die Frequenz der Untersuchungen hängt dabei vom SMA-Typ und der Ausprägung der Atemmuskulaturbeteiligung ab [11,40]:
- SMA-Typ 1 und Non-Sitter: Alle 3 Monate
- SMA-Typ 2 und 3 sowie Sitter: Alle 6 Monate
Falls erforderlich, sollte eine Überweisung in ein spezialisiertes neuromuskuläres Zentrum erfolgen, das Erfahrung mit chronischen Atemproblemen hat [11].
Da das chronische Atemmuskelversagen zunächst im Schlaf auftritt, sollten bei SMA-Typ 1 und Non-Sittern regelmäßig Symptome einer nächtlichen Hypoventilation abgefragt werden. Bei Verdacht auf eine Beeinträchtigung kann eine Blutgasanalyse durchgeführt werden. Die Vitalkapazität (VK) ist bei SMA-Typ 2 und 3 sowie Sittern ein wichtiger Indikator für mögliche Schlafatemstörungen und sollte bei jeder Untersuchung überprüft werden [11]. Falls eine akute oder chronische Atemschwäche auftritt oder es zu Atemnot kommt, kann eine nicht-invasive Beatmung (Überdruckbeatmung) helfen [11,40]. Häufig liegt zudem eine Schwäche oder Insuffizienz des Hustenstoßes vor. Daher ist ein effektives Sekretmanagement und ggf. die Versorgung mit einem Hustenassistenten essenziell für die Prophylaxe und Therapie von Atemwegsinfekten [11,40].
Kardiologie
Auch die kardiale Beteiligung ist abhängig vom SMA-Typ und umfasst häufig strukturelle Herzdefekte und kardiale Reizleitungsstörungen bei SMA-Typ 1, EKG-Veränderungen bei Typ 2 und Rhythmusstörungen bei Typ 3. Daher wird direkt nach der Diagnose eine kinderärztliche Herzuntersuchung (EKG und Echokardiographie) empfohlen, um angeborene Herzfehler oder eine Herzmuskelerkrankung auszuschließen [11,40].
Orthopädie
Unbehandelt kann eine Skoliose, die etwa 80 % der nicht gehfähigen Patient*innen mit SMA-Typ 1 oder -Typ 2 betrifft [11], zu einer fortschreitenden Wirbelsäulenverkrümmung führen, die das Sitzen erschwert, Schmerzen verursacht und die Beweglichkeit des Brustkorbs einschränkt. Letzteres kann wiederum die Lungenfunktion deutlich beeinträchtigen. Daher sind regelmäßige klinische Wirbelsäulenuntersuchungen (idealerweise alle 6 Monate) und bei Bedarf radiologische Kontrollen entscheidend für die frühzeitige Erkennung und Behandlung einer Skoliose [11]. Bereits bei ersten Anzeichen einer Verkrümmung sollte eine Vorstellung in einem auf Skoliose spezialisierten neuromuskulären Zentrum erfolgen, um eine adäquate diagnostische und therapeutische Einschätzung sicherzustellen [11].
In den aktuellen Leitlinien wird empfohlen, der Skoliose-Behandlung große Bedeutung beizumessen, die entweder konservativ oder operativ erfolgt [11,39]. Bei der konservativen Therapie kommt Physiotherapie und ein individuell angepasstes Korsett zum Einsatz. Falls eine Korsett-Therapie nicht ausreicht, können wachstumsfreundliche Wirbelsäulenimplantate eingesetzt werden. Eine Wirbelsäulenversteifung (Derotationsspondylodese) kann gegen Ende des Wachstums sinnvoll sein. Diese Operationen sollten in einem spezialisierten Zentrum für neuromuskuläre Skoliose durchgeführt werden [11].
Gastroenterologie und Ernährungsberatung
Besonders für Non-Sitter stellt die Ernährung eine Herausforderung dar. Dysphagie, Saugschwäche und die erhöhte Atemarbeit können zu Wachstumsstörungen und Unterernährung führen [14]. Daher ist eine regelmäßige Gewichtskontrolle bei Säuglingen und Kindern essenziell [39]. Zur Beurteilung der Schluckfunktion kann eine flexible endoskopische Schluckuntersuchung durchgeführt werden [11]. Falls eine ausreichende Nahrungsaufnahme nicht möglich ist, sollte eine perkutane endoskopische Gastrostomie in Erwägung gezogen werden [11,39].
Kau- und Schluckstörungen bei Non-Sittern und Sittern bergen das Risiko einer Aspiration, Atemwegsverlegung und pulmonaler Infekte. Gleichzeitig neigen Sitter und Walker aufgrund der eingeschränkten Mobilität häufiger zu Adipositas [11,40].
Die Beratung durch eine*n Ernährungsexpert*in stellt sicher, dass die Zufuhr von Makro- und Mikronährstoffen sowie Flüssigkeit bedarfsgerecht erfolgt. Besonders wichtig ist die jährliche Kontrolle des Vitamin-D3-Spiegels. Falls nötig, sollte eine Supplementierung erfolgen, um Rachitis oder Osteomalazie zu verhindern [11,39,40].
Physiotherapie, Logopädie, Ergotherapie und Hilfsmittelversorgung
Der Erhalt der Selbstständigkeit und die Förderung der Teilhabe im Alltag spielt eine zentrale Rolle bei der Behandlung der 5q-SMA [14]. Die Physiotherapie ist ein wichtiger Baustein bei der Behandlung von motorischen Einschränkungen bei der 5q-SMA mit dem Ziel, die Ausdauer und Muskelkraft zu erhalten sowie Gelenkkontrakturen hinauszuzögern [14,39]. Viele Patient*innen entwickeln Kompensationsstrategien, bei denen stärkere Muskelgruppen die Funktion schwächerer Muskeln übernehmen. Diese Strategien sollten zugelassen werden, sofern sie das Funktionsniveau stabilisieren oder verbessern [11,39].
Ergotherapie ergänzt die motorische Förderung, indem sie die Beweglichkeit, Geschicklichkeit und Selbstversorgung stärkt. Dabei geht es nicht nur um grundlegende Bewegungsfunktionen wie Stehen, Sitzen und Gehen, sondern auch um alltägliche Aktivitäten wie An- und Auskleiden, Körperpflege, Einkaufen und das Zubereiten von Mahlzeiten [11].
Besonders für Non-Sitter und Sitter ist zudem eine gezielte Atemphysiotherapie von Anfang an wichtig, um Einschränkungen frühzeitig und aktiv entgegenzuwirken. Ergänzend hilft die logopädische Therapie, auch zur Verbesserung der Beweglichkeit von Zunge und Kiefer, was sich positiv auf Sprache und Verständigung, Nahrungsaufnahme und Schlucken auswirkt [11,38].
Ein weiterer wichtiger Aspekt ist die Auswahl und Nutzung geeigneter Hilfsmittel. Wenn motorische Meilensteine aufgrund muskulärer Schwäche nicht oder erst stark verzögert erreicht werden, sollte geprüft werden, ob eine Hilfsmittelversorgung notwendig ist [11]. Dazu zählen Lagerungshilfen, Orthesen und Korsetts, Trainingsgeräte sowie Geh- und Stehhilfen [11,14,39].
Palliativversorgung und Sozialmedizin
Die 5q-SMA ist eine lebenslimitierende Erkrankung, insbesondere für Patient*innen mit SMA-Typ 0, 1 und 2, die keine oder nur eine symptomatische medikamentöse Therapie erhalten [11]. Mittelbar oder unmittelbar Betroffene sollten palliativ versorgt werden, um belastende Symptome zu lindern und die Lebensqualität unter Berücksichtigung neuer Therapie- und Versorgungsmöglichkeiten zu verbessern [11,40].
Da die Erkrankung weitreichende Auswirkungen auf den Alltag und das soziale Leben hat, ist eine ganzheitliche Betreuung essenziell [38]. Studien zeigten, dass die 5q-SMA nicht nur Alltagsaktivitäten und die soziale Teilhabe, sondern auch die psychische Verfassung der Betroffenen und ihrer Angehörigen erheblich beeinträchtigt [41]. Deshalb ist es wichtig, physische, psychische und soziale Belastungen frühzeitig zu erkennen und gezielt zu reduzieren [11]. Eine optimale Betreuung sollte in multidisziplinären Einrichtungen erfolgen, die neben der medizinischen Behandlung auch psychosoziale Aspekte einbeziehen [38]. Eine Sozialberatung ist sehr wichtig, um den SMA-Patient*innen Zugang zu Hilfsangeboten zu ermöglichen.
Fazit
Die 5q-SMA ist eine seltene, autosomal-rezessiv vererbte neurodegenerative Erkrankung, deren Symptomatik und Krankheitsverlauf je nach SMA-Typ stark variiert. Typischerweise äußert sich die Erkrankung durch generalisierte Muskelschwäche, die bei den am schwersten betroffenen Patient*innen des SMA-Typ 0 unbehandelt im Alter von 6 Monaten zum Tod führt. Eine frühzeitige Diagnosestellung – idealerweise noch in der präsymptomatischen Phase – ist entscheidend für den Therapieerfolg, um das Fortschreiten der Erkrankung zu verlangsamen oder zu stoppen. In Deutschland stehen derzeit drei für SMA zugelassene Therapien, Nusinersen, Risdiplam und Onasemnogen Abeparvovec , zur Verfügung. Die Versorgung dieser komplexen Erkrankung sollte idealerweise in spezialisierten neuromuskulären Zentren erfolgen, die eine umfassende, eng abgestimmte interdisziplinäre Betreuung durch die Fachbereiche Neurologie, Orthopädie, Pneumologie, Physiotherapie und Sozialmedizin bieten. Dank ihrer speziellen Expertise in der Behandlung der 5q-SMA gewährleisten neuromuskuläre Zentren eine optimale Versorgung durch individuelle Therapieplanung, kontinuierliche klinische Verlaufsbeurteilung, sowie die gezielte Behandlung krankheitsspezifischer Symptome. Ziel ist es, Patient*innen und ihre Angehörigen im Alltag bestmöglich zu unterstützen und die Lebensqualität langfristig zu verbessern.
Literatur
1. Elsheikh BH, Zhang X, Swoboda KJ, et al. ‚Pregnancy and delivery in women with spinal muscular atrophy‘. Int J Neurosci. 2017;127(11):953-7.
2. Faravelli I, Nizzardo M, Comi GP, et al. ‚Spinal muscular atrophy–recent therapeutic advances for an old challenge‘. Nat Rev Neurol. 2015;11(6):351-9.
3. Vill K, Kölbel H, Schwartz O, et al. ‚One Year of Newborn Screening for SMA – Results of a German Pilot Project‘. J Neuromuscul Dis. 2019;6(4):503-15.
4. Wirth B, Karakaya M, Kye MJ, et al. ‚Twenty-Five Years of Spinal Muscular Atrophy Research: From Phenotype to Genotype to Therapy, and What Comes Next‘. Annu Rev Genomics Hum Genet. 2020;21:231-61.
5. d’Ydewalle C, Sumner CJ. ‚Spinal Muscular Atrophy Therapeutics: Where do we Stand?‘. Neurotherapeutics. 2015;12(2):303-16.
6. Wang CH, Lunn MR. ‚Spinal muscular atrophy: advances in research and consensus on care of patients‘. Curr Treat Options Neurol. 2008;10(6):420-8.
7. Cordts I, Lingor P, Friedrich B, et al. ‚Intrathecal nusinersen administration in adult spinal muscular atrophy patients with complex spinal anatomy‘. Ther Adv Neurol Disord. 2020;13:1756286419887616.
8. Fachinformation Nusinersen 2024 [Verfügbar unter: , Stand Januar 2025]. Letzter Zugriff: 12.06.2025.
9. Fachinformation Risdiplam 2024 [Verfügbar unter: , Stand Juni 2025]. Letzter Zugriff: 12.06.2025.
10. Fachinformation Onasemnogen Abeparvovec 2024 [Verfügbar unter: , Stand März 2025]. Letzter Zugriff: 12.06.2025.
11. AWMF S2k-Leitlinie: Diagnostik und Therapie der 5q-assoziierten spinalen Muskelatrophie im Kindes- und Erwachsenenalter 2025 [Verfügbar unter: ].
12. Day JW, Howell K, Place A, et al. ‚Advances and limitations for the treatment of spinal muscular atrophy‘. BMC Pediatr. 2022;22(1):632.
13. Wang CH, Finkel RS, Bertini ES, et al. ‚Consensus statement for standard of care in spinal muscular atrophy‘. J Child Neurol. 2007;22(8):1027-49.
14. Arnold WD, Kassar D, Kissel JT. ‚Spinal muscular atrophy: diagnosis and management in a new therapeutic era‘. Muscle Nerve. 2015;51(2):157-67.
15. Darras BT. ‚Spinal muscular atrophies‘. Pediatr Clin North Am. 2015;62(3):743-66.
16. Shanmugarajan S, Swoboda KJ, Iannaccone ST, et al. ‚Congenital bone fractures in spinal muscular atrophy: functional role for SMN protein in bone remodeling‘. J Child Neurol. 2007;22(8):967-73.
17. D’Amico A, Mercuri E, Tiziano FD, et al. ‚Spinal muscular atrophy‘. Orphanet J Rare Dis. 2011;6:71.
18. Lunn MR, Wang CH. ‚Spinal muscular atrophy‘. Lancet. 2008;371(9630):2120-33.
19. Finkel RS, Mercuri E, Darras BT, et al. ‚Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy‘. N Engl J Med. 2017;377(18):1723-32.
20. Mercuri E, Darras BT, Chiriboga CA, et al. ‚Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy‘. N Engl J Med. 2018;378(7):625-35.
21. Günther R, Wurster CD, Brakemeier S, et al. ‚Long-term efficacy and safety of nusinersen in adults with 5q spinal muscular atrophy: a prospective European multinational observational study‘. Lancet Reg Health Eur. 2024;39:100862.
22. Masson R, Mazurkiewicz-Bełdzińska M, Rose K, et al. ‚Safety and efficacy of risdiplam in patients with type 1 spinal muscular atrophy (FIREFISH part 2): secondary analyses from an open-label trial‘. Lancet Neurol. 2022;21(12):1110-9.
23. Mercuri E, Deconinck N, Mazzone ES, et al. ‚Safety and efficacy of once-daily risdiplam in type 2 and non-ambulant type 3 spinal muscular atrophy (SUNFISH part 2): a phase 3, double-blind, randomised, placebo-controlled trial‘. Lancet Neurol. 2022;21(1):42-52.
24. McMillan HJ, Baranello G, Farrar MA, et al. ‚Safety and Efficacy of IV Onasemnogene Abeparvovec for Pediatric Patients With Spinal Muscular Atrophy: The Phase 3b SMART Study‘. Neurology. 2025;104(2):e210268.
25. Gowda V, Atherton M, Murugan A, et al. ‚Efficacy and safety of onasemnogene abeparvovec in children with spinal muscular atrophy type 1: real-world evidence from 6 infusion centres in the United Kingdom‘. The Lancet Regional Health – Europe. 2024;37.
26. Kirschner J, Bernert G, Butoianu N, et al. ‚2024 update: European consensus statement on gene therapy for spinal muscular atrophy‘. Eur J Paediatr Neurol. 2024;51:73-8.
27. ‚WHO Motor Development Study: windows of achievement for six gross motor development milestones‘. Acta Paediatr Suppl. 2006;450:86-95.
28. Ramsey D, Scoto M, Mayhew A, et al. ‚Revised Hammersmith Scale for spinal muscular atrophy: A SMA specific clinical outcome assessment tool‘. PLoS One. 2017;12(2):e0172346.
29. Cano SJ, Mayhew A, Glanzman AM, et al. ‚Rasch analysis of clinical outcome measures in spinal muscular atrophy‘. Muscle Nerve. 2014;49(3):422-30.
30. Glanzman AM, Mazzone E, Main M, et al. ‚The Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND): test development and reliability‘. Neuromuscul Disord. 2010;20(3):155-61.
31. Mazzone E, Bianco F, Martinelli D, et al. ‚Assessing upper limb function in nonambulant SMA patients: development of a new module‘. Neuromuscul Disord. 2011;21(6):406-12.
32. Main M, Kairon H, Mercuri E, et al. ‚The Hammersmith functional motor scale for children with spinal muscular atrophy: a scale to test ability and monitor progress in children with limited ambulation‘. Eur J Paediatr Neurol. 2003;7(4):155-9.
33. Montes J, McDermott MP, Martens WB, et al. ‚Six-Minute Walk Test demonstrates motor fatigue in spinal muscular atrophy‘. Neurology. 2010;74(10):833-8.
34. Pechmann A, König K, Bernert G, et al. ‚SMArtCARE – A platform to collect real-life outcome data of patients with spinal muscular atrophy‘. Orphanet J Rare Dis. 2019;14(1):18.
35. DRKS – Deutsches Register Klinischer Studien: Longitudinale Datensammlung zum Krankheitsverlauf von Patienten mit Spinaler Muskelatrophie: Die SMArtCARE Datenbank 2018 [Verfügbar unter: ]. Letzter Zugriff: 25.06.2025.
36. Gemeinsamer Bundesausschuss: Anwendungsbegleitende Datenerhebung und Beschränkung der Versorgungsbefugnis: Onasemnogen-Abeparvovec – Spinale Muskelatrophie 2022 [Verfügbar unter: ]. Letzter Zugriff: 25.06.2025.
37. Gemeinsamer Bundesausschuss: Anwendungsbegleitende Datenerhebung zu Risdiplam startet am 30. Oktober 2024 [Verfügbar unter: ]. Letzter Zugriff: 25.06.2025.
38. Ropars J, Peudenier S, Genot A, et al. ‚Multidisciplinary approach and psychosocial management of spinal muscular atrophy (SMA)‘. Arch Pediatr. 2020;27(7s):7s45-7s9.
39. Mercuri E, Finkel RS, Muntoni F, et al. ‚Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care‘. Neuromuscul Disord. 2018;28(2):103-15.
40. Finkel RS, Mercuri E, Meyer OH, et al. ‚Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics‘. Neuromuscul Disord. 2018;28(3):197-207.
41. Qian Y, McGraw S, Henne J, et al. ‚Understanding the experiences and needs of individuals with Spinal Muscular Atrophy and their parents: a qualitative study‘. BMC Neurol. 2015;15:217.
Tutorielle Unterstützung
Die tutorielle Unterstützung der Fortbildungsteilnehmer erfolgt durch unseren ärztlichen Leiter Dr. med. Alexander Voigt in Zusammenarbeit mit der arztCME-Redaktion. Inhaltliche Fragen können über das Kommentarfeld, direkt per Mail an service@arztcme.de oder via Telefon unter Tel.: +49(0)180-3000759 gestellt werden. Inhaltliche Fragen werden von unserem ärztlichen Leiter bzw. nach Rücksprache mit diesem und evtl. dem Autor auch von der arztCME-Redaktion beantwortet.
Technischer Support
Der technische Support der Online-Akademie arztCME.de erfolgt durch geschulte Mitarbeiterinnen und Mitarbeiter des Betreibers health&media GmbH unter der E-Mail-Adresse technik@arztcme.de oder via Telefon unter Tel.: 49(0)180-3000759.