Kardiale Beteiligung bei der Friedreich-Ataxie
Interessengebiete: Allgemeinmedizin und Innere Medizin, Neurologie, Kardiologie
Friedreich-Ataxie (FA) ist die häufigste vererbte Ataxie in Europa und eine multisystemische, autosomal-rezessive neurodegenerative Erkrankung. Neben den klassischen neurologischen Symptomen ist insbesondere die Herzbeteiligung ausschlaggebend für Prognose und Lebenserwartung der Betroffenen. Die kardiale Beteiligung wird häufig spät erkannt, was die Behandlung erschwert und die Lebensqualität einschränkt. Deshalb sind frühzeitige Diagnostik, regelmäßige Überwachung und leitliniengerechte Therapie essenziell. Diese Fortbildung vermittelt praxisnah die pathophysiologischen Grundlagen, diagnostischen Möglichkeiten, die stadiengerechte Beurteilung sowie therapeutische Ansätze. Zudem wird die Bedeutung der interdisziplinären Zusammenarbeit zwischen Kardiologie und Neurologie hervorgehoben.

Kursinhalt
Inhaltsverzeichnis
- Einleitung
- Kardiale Manifestationen
- Pathophysiologie der Kardiomyopathie bei FA
- Diagnostik
- Elektrokardiogramm (EKG)
- Echokardiographie
- Kardiale Magnetresonanztomographie (cMRT)
- Biomarker
- Stadieneinteilung der kardialen Beteiligung bei Friedreich-Ataxie
- Klinisches Vorgehen und leitliniengerechtes Monitoring
- Therapie
- Fazit
- Literatur
- Bildquellen
Einleitung
Die Friedreich-Ataxie (FA) ist eine autosomal-rezessiv vererbte, progressive neurodegenerative Erkrankung und die häufigste erbliche Form der Ataxie [1, 2]. Der Krankheitsbeginn liegt typischerweise im Jugendalter zwischen dem 10. und 15. Lebensjahr. In Deutschland beträgt die Prävalenz etwa 3–4 Fälle pro 100.000 Personen [1, 3].
Ursächlich ist in der Mehrzahl der Fälle eine GAA-Trinukleotid-Repeat-Expansion im Frataxin-Gen, das für das mitochondriale Protein Frataxin kodiert [4]. Der resultierende Frataxinmangel verursacht eine fortschreitende Zellschädigung in besonders energieabhängigen Geweben, was die FA zu einer ausgeprägt multisystemischen Erkrankung macht [5].
Neben dem Nervensystem sind insbesondere das Herz-, Muskel-Skelett- und endokrine System betroffen [1]. Neurologische und neuromuskuläre Symptome umfassen Gang- und Standataxie, Verlust der Tiefensensibilität, Spastiken und Dysarthrie. Zudem treten häufig Skelettveränderungen wie Skoliose oder Fußdeformitäten und Diabetes mellitus auf [1, 6].
Im Verlauf kommt es zu erheblichen Einschränkungen der Mobilität und Lebensqualität, sodass die meisten Betroffenen innerhalb von 10 bis 20 Jahren nach Krankheitsbeginn auf einen Rollstuhl angewiesen sind [7]. Die durchschnittliche Lebenserwartung beträgt bei FA lediglich etwa 37 Jahre [7, 8]. Hauptursache der vorzeitigen Mortalität ist die kardiale Beteiligung [2].
Kardiale Manifestationen
Eine kardiale Beteiligung tritt im Krankheitsverlauf bei nahezu allen Patientinnen und Patienten mit FA auf. Etwa zwei Drittel der Betroffenen entwickeln eine hypertrophe Kardiomyopathie, meist in Form einer linksventrikulären hypertrophen Kardiomyopathie [9-11]. Im weiteren Verlauf kann dies in eine dilatative Kardiomyopathie übergehen [6, 9, 11].
Die fortschreitende myokardiale Hypertrophie führt zu einer reduzierten Perfusionsreserve und letztlich zur Herzinsuffizienz [9, 11]. Im Rahmen der dilatativen Kardiomyopathie können häufig supraventrikulär Arrhythmien, insbesondere Vorhofflimmern auftreten [Abb. 1] [6, 11].
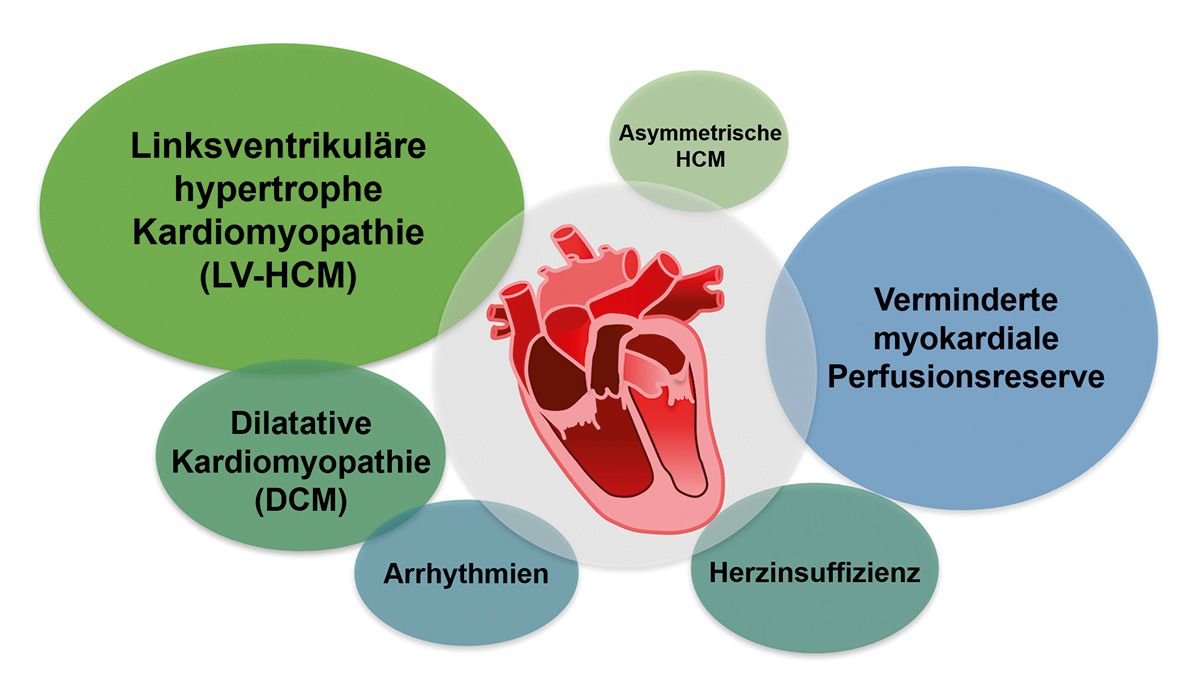
Abb. 1: Formen der kardialen Beteiligung bei FA [6]
Auffällig ist, dass schwere kardiale Manifestationen tendenziell häufiger bei jüngeren FA-Betroffenen auftreten [2, 12]. Etwa die Hälfte der unter 20-Jährigen weist eine mittelgradige oder schwere Kardiomyopathie auf, während dies bei den 40-50-Jährigen lediglich bei etwa 9 % der Fall ist [12]. Zwischen dem Schweregrad der neurologischen Symptome und dem Ausmaß der Kardiomyopathie besteht jedoch kein direkter Zusammenhang [12]. Allerdings wurde gezeigt, dass die Ausprägung der Hypertrophie mit der Anzahl der GAA-Repeat-Wiederholungen zunimmt [13, 14].
Pathophysiologie der Kardiomyopathie bei FA
Das Frataxin-Protein spielt eine zentrale Rolle im mitochondrialen Eisentransport. Bei der FA führt der Mangel an Frataxin zu einer Störung der mitochondrialen Eisenhomöostase [2, 10]. Da Mitochondrien die Hauptquelle der zellulären Energieproduktion sind, sind insbesondere energieabhängige Organe wie das Herz und das Nervensystem von diesem Defekt betroffen [5, 15].
Im Myokard führt der Frataxin-Mangel zu Eisenablagerungen in den Mitochondrien und im Zytoplasma, einer reduzierten Energiegewinnung sowie oxidativem Stress infolge der vermehrten Bildung reaktiver Sauerstoffspezies. Diese Prozesse wiederum lösen entzündliche Reaktionen und Zelluntergang [Apoptose und Nekrose] im Herzmuskel aus [2, 10].
Der Verlust kontraktiler Kardiomyozyten wird zunächst durch kompensatorische Hypertrophie der verbleibenden Herzmuskelzellen ausgeglichen, was zu einer zunehmenden Wandverdickung führt [10]. Mit fortschreitender Erkrankung entwickelt sich eine Fibrosierung des Myokards, bei der die abgestorbenen Myozyten zunehmend durch Fibrozyten ersetzt werden [2]. Dieser Remodeling-Prozess führt zu einer steifen, fibrotischen Herzwand mit eingeschränkter diastolischer Funktion. Im weiteren Verlauf führt der Verlust der myokardialen Elastizität zu einer konsekutiven Dilatation, die mit einer systolischen Dysfunktion einhergeht. Die im Rahmen der Fibrosierung entstehenden elektrischen Leitungsstörungen begünstigen zudem das Auftreten von Arrhythmien [10, 16].
Klinische Bedeutung der Herzbeteiligung bei FA
Die Herzbeteiligung ist die häufigste Todesursache bei FA und verantwortlich für die deutlich verkürzte Lebenserwartung – im Mittel beträgt sie nur etwa die Hälfte der normalen Lebenserwartung [6, 17, 18]. Während in der Allgemeinbevölkerung rund 40 % der Todesfälle kardial bedingt sind [19], liegt dieser Anteil bei Personen mit FA bei etwa 60 % [6, 10]. Obwohl die genauen Mechanismen noch nicht vollständig geklärt sind, gelten Rhythmusstörungen und Herzinsuffizienz als die Hauptursachen des vorzeitigen Versterbens [2].
Eine frühzeitige Diagnose der kardialen Beteiligung ist daher entscheidend für die Prognose. Die Diagnosestellung ist jedoch oft erschwert, da klinische Symptome lange fehlen [6]. Typisch für einen Verdacht auf kardiale Erkrankungen sind verminderte Belastbarkeit der Patientinnen und Patienten im Alltag, wie etwa beim Treppensteigen. Bei FA reduziert die neuromuskuläre Einschränkung allerdings die körperliche Belastbarkeit, sodass Belastungsdyspnoe oder Leistungsabfall häufig erst in fortgeschrittenen Krankheitsstadien auffallen [6, 12].
Diagnostik
Eine regelmäßige kardiologische Verlaufskontrolle ist bei Patientinnen und Patienten mit FA unverzichtbar, auch wenn zunächst keine Symptome bestehen. Zur Beurteilung einer kardialen Beteiligung stehen dabei verschiedene diagnostische Verfahren zur Verfügung, die charakteristische Befunde und Funktionsveränderungen des Herzens bei FA aufzeigen können (Abb. 2) [11].

Abb. 2: Diagnostische Methoden zum Nachweis einer kardialen Beteiligung bei FA [11]
Elektrokardiogramm (EKG)
Das EKG ist meist bereits in der hausärztlichen Versorgung verfügbar und ermöglicht die Beurteilung der elektrischen Erregungsleitung und -rückbildung des Herzens sowie die Erkennung von Rhythmusstörungen [11].
Typische EKG-Befunde bei FA sind T-Wellen-Inversionen in den inferioren und lateralen Ableitungen (II, III, aVF, V4–V6) [20]. Diese gelten als frühes Zeichen einer kardialen Beteiligung und sind bei fast allen FA-Betroffenen mit einer Kardiomyopathie vorhanden [11, 20]. Im fortgeschrittenen Stadium zeigen sich häufig Zeichen einer linksventrikulären Hypertrophie – typischerweise tiefe S-Zacken in V1/V2 und hohe R-Zacken in V5/V6 [16]. Als Richtwert gilt bei Erwachsenen eine Summenamplitude von R- und S-Zacke über 3,5 mV. Eine Vergrößerung dieser Summenamplitude weist auf eine Hypertrophie hin.
Echokardiographie
Die Echokardiographie ist das zentrale Instrument in der Diagnostik der kardialen Beteiligung bei FA. Sie ermöglicht die Beurteilung von Wanddicke, Kammergröße, systolischer und diastolischer Funktion [11].
Typischerweise findet sich bei FA eine konzentrische linksventrikuläre Hypertrophie mit einer Wanddicke von <15 mm (Abb. 3 A) [16]. Eine Obstruktion des linksventrikulären Ausflusstrakts tritt dagegen nur selten auf [16].
Im Vier-Kammer-Blick zeigt sich im weiteren Verlauf eine Dilatation des linken Ventrikels sowie eine diastolische Funktionsstörung, die lange Zeit als pseudonormal erscheinen kann [17, 21] (Abb. 3 B). Mit fortschreitender Erkrankung kommt es durch zunehmende Myokardfibrose zu einer Abnahme der Ejektionsfraktion und somit zur Entwicklung einer systolischen Dysfunktion [16].
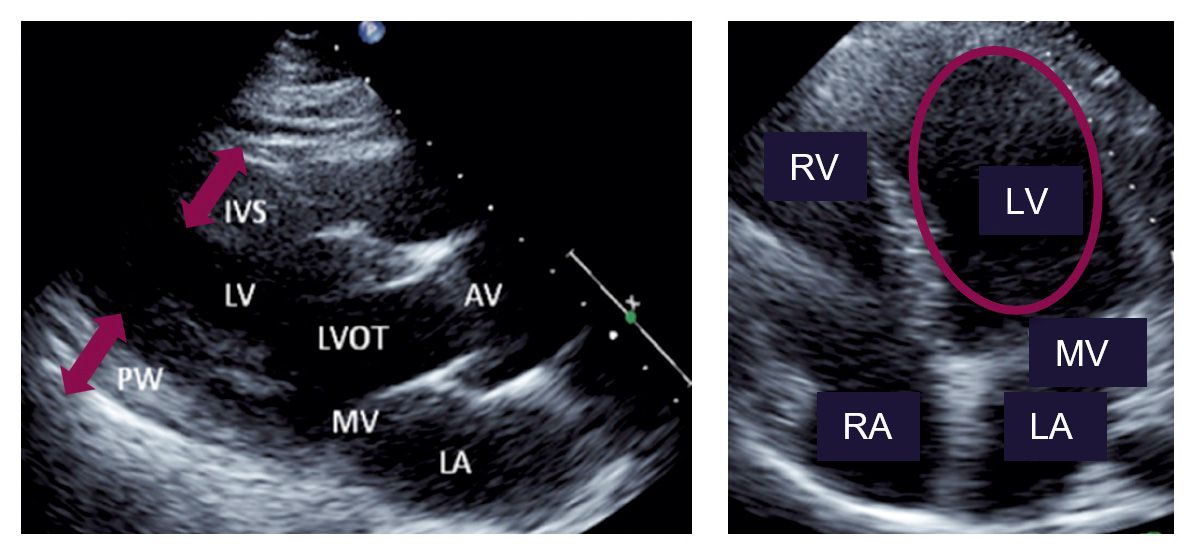
Abb. 3: Echokardiogramme bei FA. A) Vergrößerung des interventrikulären Septums (IVS) und der posterioren Hinterwand (PW). B) Dilatation des linken Ventrikels (LV) [6]
Kardiale Magnetresonanztomographie (cMRT)
Das kardiale MRT bietet eine besonders präzise Quantifizierung von Hypertrophie, Funktionsparametern und Gewebecharakterisierung [11]. Es kann insbesondere bei schwereren Formen der kardialen Beteiligung mittels Late Gadolinium Enhancement (LGE) helfen, das Ausmaß der Myokardfibrose zu ermitteln.
Fibrotisches Gewebe zeigt eine verzögerte Kontrastmittel-Rückbildung und erscheint als hell aufleuchtende Areale im MRT. Typischerweise findet sich bei Personen mit FA eine fleckige, unregelmäßige Fibrose, vor allem im linksventrikulären Septum und an der posterolateralen Wand (Abb. 4) [11].
Im Endstadium weisen nahezu alle Betroffenen ausgedehnte Myokardfibrosen auf, die mit einer deutlichen Einschränkung der Herzfunktion einhergehen [11, 20].
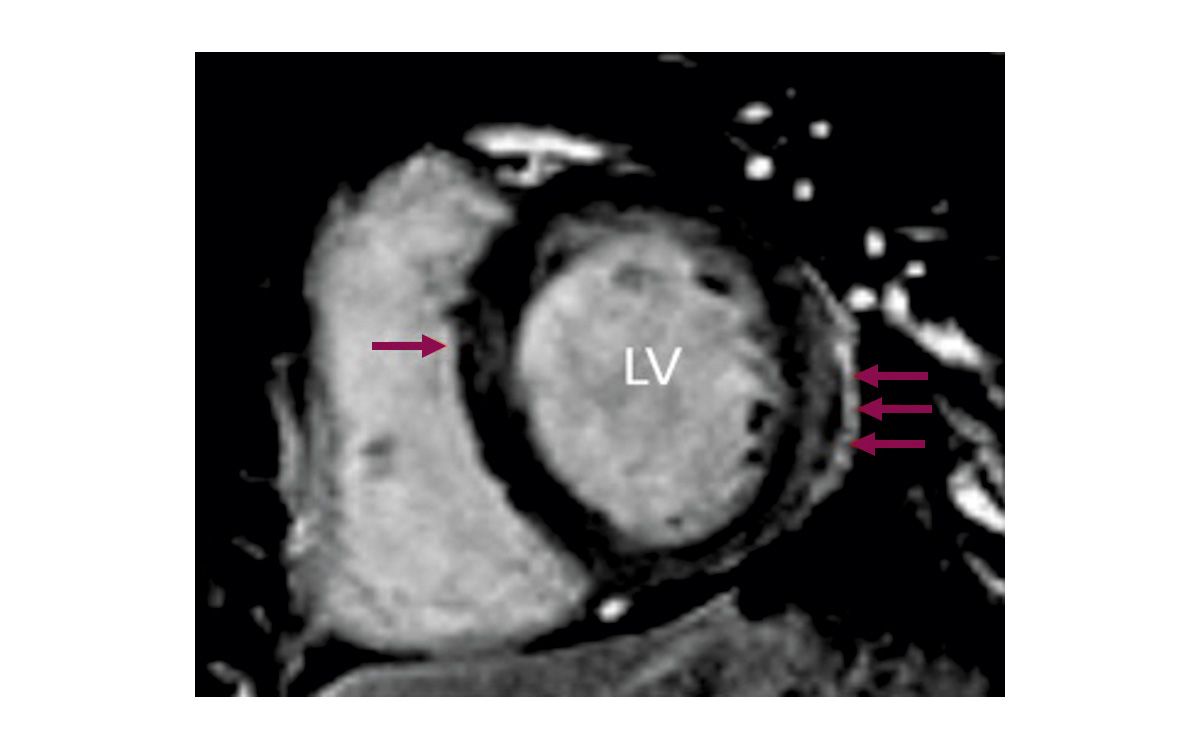
Abb. 4. Unregelmäßige Fibrose des linken Ventrikels (LV) [17]
Ein Adenosin-Stress-MRT kann zusätzlich zur Beurteilung der myokardialen Perfusionsreserve eingesetzt werden. Unter Adenosin-Stimulation – die eine erhöhte Koronardurchblutung provoziert – zeigt sich bei vielen FA-Betroffenen eine verminderte Perfusionsreserve, oft bereits vor dem Auftreten klinischer Symptome [16, 22].
Biomarker
Zur Beurteilung einer myokardialen Schädigung können neben bildgebenden Verfahren auch Serumbiomarker herangezogen werden.
Bei einer Schädigung oder Nekrose von Herzmuskelzellen werden kardiales hochsensitives Troponin T (hsTnT) und Troponin I (cTnI) ins Blut freigesetzt [23]. Diese Marker dienen allgemein zur Diagnostik von Kardiomyopathien und werden auch bei der FA eingesetzt, um eine subklinische myokardiale Beteiligung nachzuweisen [20, 23]. Erhöhte Troponinwerte können insbesondere auf eine Verdickung der Herzscheidewand (septale Hypertrophie) hinweisen. Die Normwerte liegen bei hsTnT < 14 ng/l und cTnI 30–100 ng/l [23].
Diese Grenzwerte werden bei über 30 % der Menschen mit FA überschritten – häufig bereits vor dem Auftreten struktureller Veränderungen im Herzmuskel [23, 24]. Zudem zeigen etwa 70 % der Patientinnen und Patienten mit kardialen Ereignissen erhöhte Troponinwerte, was ihre diagnostische und prognostische Relevanz unterstreicht [20, 24].
Ein weiterer relevanter Biomarker ist das N-terminale pro-B-Typ natriuretische Peptid (NT-proBNP). Erhöhte NT-proBNP-Spiegel deuten auf einen erhöhten myokardialen Füllungsdruck und somit auf eine drohende Herzinsuffizienz hin [23]. Etwa 14 % der Personen mit FA zeigen NT-proBNP-Konzentrationen über dem Normbereich von 100–125 ng/l [23]. Bei Menschen mit vorbestehender Herzinsuffizienz oder arrhythmischen Ereignissen können die Werte sogar bis zu 775 ng/l erreichen [23].
Stadieneinteilung der kardialen Beteiligung bei Friedreich-Ataxie
Weidemann et al. haben ein neues Stagingmodell zur Beurteilung der myokardialen Beteiligung bei FA vorgeschlagen [20]. Dieses Modell basiert auf einer Kombination klinischer, bildgebender und laborchemischer Parameter und ermöglicht eine standardisierte Einordnung des Schweregrads der Kardiomyopathie (Abb. 5).
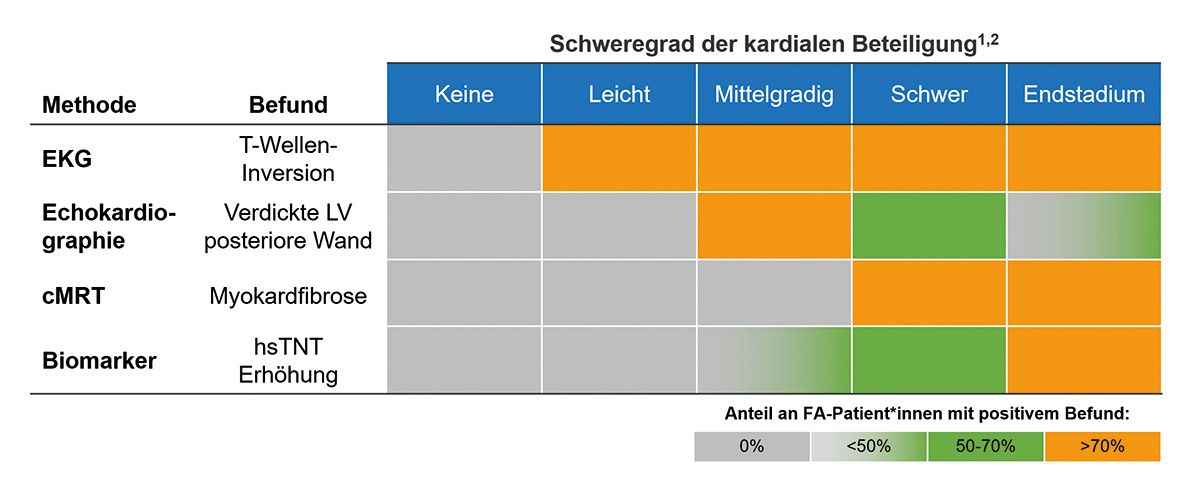
Abb. 5: Beurteilung und Einteilung der kardialen Beteiligung bei FA [20]. Das Farbschema zeigt die Anteile der Patientinnen und Patienten mit positivem Befund im jeweiligen Stadium [20].
Charakteristisch für Patienten und Patientinnen im Endstadium der Kardiomyopathie ist zudem eine deutlich reduzierte linksventrikuläre Ejektionsfraktion (<55 %) [20].
Dieses Staging-System bietet eine praxisnahe und pathophysiologisch fundierte Grundlage, um die kardiale Beteiligung bei FA systematisch zu erfassen, den Krankheitsverlauf zu überwachen und Therapieentscheidungen gezielter zu treffen [20].
Klinisches Vorgehen und leitliniengerechtes Monitoring
Die Betreuung von Patientinnen und Patienten mit FA sollte interdisziplinär erfolgen, in enger Zusammenarbeit zwischen Neurologen und Kardiologen. Bereits bei Diagnosestellung ist eine kardiologische Mitbetreuung sinnvoll, um kardiale Veränderungen frühzeitig zu erkennen und zu überwachen. Empfohlen werden regelmäßige Verlaufskontrollen mit Ruhe-EKG und Echokardiographie mindestens einmal jährlich, um das Auftreten oder Fortschreiten einer kardialen Beteiligung frühzeitig zu erfassen [8, 25].
Bei Verdacht auf Rhythmusstörungen – etwa supraventrikuläre oder ventrikuläre Extrasystolen – sollte ein Langzeit-EKG durchgeführt werden, insbesondere bei Palpitationen, Synkopen oder einem auffälligen Basis-EKG [8]. Moderne Wearables, wie Smartwatches mit integrierter 1-Kanal-EKG-Funktion, können eine ergänzende Überwachung ermöglichen und bei der Diagnosestellung unterstützen.
Eine kardiologische Untersuchung ist in jedem Fall angezeigt bei klinischen Symptomen, auffälligen EKG-Befunden oder pathologisch erhöhten Biomarkern (z. B. Troponin, NT-proBNP). Vor größeren chirurgischen Eingriffen, insbesondere unter Allgemeinanästhesie, ist eine kardiologische Beurteilung essenziell, da unerkannte Herzinsuffizienzen oder Arrhythmien das perioperative Risiko deutlich erhöhen können [8].
Therapie
Seit 2024 steht mit Omaveloxolon erstmals eine kausal orientierte Therapie zur Behandlung der FA für Patientinnen und Patienten ab 16 Jahren zur Verfügung. In den Zulassungsstudien konnte gezeigt werden, dass die Behandlung mit Omaveloxolon die neurologische Funktion stabilisieren oder sogar verbessern kann [26].
Die Behandlung der kardialen Manifestationen bei FA orientiert sich an den allgemeinen Leitlinien der Deutschen Gesellschaft für Kardiologie (DGK) und der European Society of Cardiology (ESC) [8, 11, 27].
Arrhythmien
Dabei sollten persistierendes oder episodisches Vorhofflimmern gemäß Standardtherapie behandelt werden. Neben Rhythmus- oder Frequenzkontrolle sollte die Behandlung mit Antikoagulation und Katheterablation erfolgen [8, 11, 25, 28]. Bei ventrikulären Arrhythmien sind Betablocker Mittel der Wahl, während antiarrhythmische Substanzen mit negativer Inotropie (außer Betablocker) vermieden werden sollten, um eine Verschlechterung der Herzschwäche zu vermeiden [8, 28].
Herzinsuffizienz
Bei asymptomatischer systolischer Dysfunktion erfolgt die leitliniengerechte Therapie mit ACE-Hemmern, Aldosteronantagonist, Angiotensinrezeptorblockern (ARB) oder Betablockern [8, 27]. Bei weiterer Einschränkung und symptomatischer Herzinsuffizienz sollte zusätzlich mit Diuretika therapiert werden [27]. Ein Einsatz von Sacubitril/Valsartan kann ebenfalls bei symptomatischer Herzinsuffizienz erwogen werden.
Bei persistierender Einschränkung (Ejektionsfraktion <35 %) trotz optimaler Therapie ist ein implantierbarer Kardioverter-Defibrillator (ICD) zur Prävention des plötzlichen Herztods zu erwägen [8, 25, 27]. Diese Entscheidung sollte jedoch individuell und unter Berücksichtigung der Lebensqualität und -erwartung gemeinsam mit den Betroffenen getroffen werden.
Eine Herztransplantation ist prinzipiell möglich, wird jedoch aufgrund der Systemerkrankung und der begrenzten Organverfügbarkeit kritisch diskutiert [2, 8, 25]. In der Regel erhalten Patientinnen und Patienten ohne systemische Grunderkrankungen Priorität [27].
Fazit
Über 60 % der Patientinnen und Patienten mit FA entwickeln im Verlauf eine kardiale Beteiligung, meist in Form einer hypertrophen Kardiomyopathie. Dabei ist die kardiale Beteiligung Haupttodesursache bei FA. Eine frühzeitige Identifikation der kardialen Beteiligung ist daher entscheidend, um die Prognose der Betroffenen zu verbessern.
Ein EKG und Echokardiogramm sollten nach Diagnosestellung und einmal im Jahr angewendet werden, während eine kardiale MRT bei fortgeschrittenen Stadien der kardialen Beteiligung herangezogen werden sollte. Die Therapie richtet sich nach etablierten Herzinsuffizienz- und Arrhythmieleitlinien, unter besonderer Berücksichtigung der individuellen Lebenssituation der Betroffenen.
Durch regelmäßige Kontrollen, rechtzeitige Diagnosestellung und leitliniengerechte Therapie können Herzinsuffizienz und Rhythmusstörungen frühzeitig erkannt und behandelt werden. Eine interdisziplinäre Zusammenarbeit zwischen Neurologie, Kardiologie und ggf. Pädiatrie ist hierbei essenziell.
Literatur
1. Cook A, Giunti P. Friedreich’s ataxia: clinical features, pathogenesis and management. British medical bulletin. 2017;124(1):19-30.
2. Hanson E, Sheldon M, Pacheco B, Alkubeysi M, Raizada V. Heart disease in Friedreich’s ataxia. World journal of cardiology. 2019;11(1):1-12.
3. Schulz JB, Boesch S, Bürk K, Dürr A, Giunti P, Mariotti C, et al. Diagnosis and treatment of Friedreich ataxia: a European perspective. Nature reviews Neurology. 2009;5(4):222-34.
4. Campuzano V, Montermini L, Moltò MD, Pianese L, Cossée M, Cavalcanti F, et al. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science (New York, NY). 1996;271(5254):1423-7.
5. Chiang S, Kalinowski DS, Jansson PJ, Richardson DR, Huang ML. Mitochondrial dysfunction in the neuro-degenerative and cardio-degenerative disease, Friedreich’s ataxia. Neurochemistry international. 2018;117:35-48.
6. Bourke T, Keane D. Friedreich’s Ataxia: a review from a cardiology perspective. Irish journal of medical science. 2011;180(4):799-805.
7. Fogel BL, Perlman S. Clinical features and molecular genetics of autosomal recessive cerebellar ataxias. The Lancet Neurology. 2007;6(3):245-57.
8. Corben LA, Collins V, Milne S, Farmer J, Musheno A, Lynch D, et al. Clinical management guidelines for Friedreich ataxia: best practice in rare diseases. Orphanet journal of rare diseases. 2022;17(1):415.
9. Legrand L, Weinsaft JW, Pousset F, Ewenczyk C, Charles P, Hatem S, et al. Characterizing cardiac phenotype in Friedreich’s ataxia: The CARFA study. Archives of cardiovascular diseases. 2022;115(1):17-28.
10. Payne RM. Cardiovascular Research in Friedreich Ataxia: Unmet Needs and Opportunities. JACC Basic to translational science. 2022;7(12):1267-83.
11. Monda E, Lioncino M, Rubino M, Passantino S, Verrillo F, Caiazza M, et al. Diagnosis and Management of Cardiovascular Involvement in Friedreich Ataxia. Heart failure clinics. 2022;18(1):31-7.
12. Weidemann F, Rummey C, Bijnens B, Störk S, Jasaityte R, Dhooge J, et al. The heart in Friedreich ataxia: definition of cardiomyopathy, disease severity, and correlation with neurological symptoms. Circulation. 2012;125(13):1626-34.
13. Rajagopalan B, Francis JM, Cooke F, Korlipara LV, Blamire AM, Schapira AH, et al. Analysis of the factors influencing the cardiac phenotype in Friedreich’s ataxia. Movement disorders : official journal of the Movement Disorder Society. 2010;25(7):846-52.
14. Legrand L, Diallo A, Monin ML, Ewenczyk C, Charles P, Isnard R, et al. Predictors of Left Ventricular Dysfunction in Friedreich’s Ataxia in a 16-Year Observational Study. American journal of cardiovascular drugs : drugs, devices, and other interventions. 2020;20(2):209-16.
15. Doni D, Cavion F, Bortolus M, Baschiera E, Muccioli S, Tombesi G, et al. Human frataxin, the Friedreich ataxia deficient protein, interacts with mitochondrial respiratory chain. Cell death & disease. 2023;14(12):805.
16. Weidemann F, Störk S, Liu D, Hu K, Herrmann S, Ertl G, et al. Cardiomyopathy of Friedreich ataxia. Journal of neurochemistry. 2013;126 Suppl 1:88-93.
17. Weidemann F, Scholz F, Florescu C, Liu D, Hu K, Herrmann S, et al. (Heart involvement in Friedreich’s ataxia). Herz. 2015;40 Suppl 1:85-90.
18. Statista. Durchschnittliche Lebenserwartung in Deutschland laut der Sterbetafel 2020/2024 2023.
19. RKI. Herz-Kreislauf-Erkrankungen 2025.
20. Weidemann F, Liu D, Hu K, Florescu C, Niemann M, Herrmann S, et al. The cardiomyopathy in Friedreich’s ataxia – New biomarker for staging cardiac involvement. International journal of cardiology. 2015;194:50-7.
21. Regner SR, Lagedrost SJ, Plappert T, Paulsen EK, Friedman LS, Snyder ML, et al. Analysis of echocardiograms in a large heterogeneous cohort of patients with friedreich ataxia. The American journal of cardiology. 2012;109(3):401-5.
22. Raman SV, Phatak K, Hoyle JC, Pennell ML, McCarthy B, Tran T, et al. Impaired myocardial perfusion reserve and fibrosis in Friedreich ataxia: a mitochondrial cardiomyopathy with metabolic syndrome. European heart journal. 2011;32(5):561-7.
23. Legrand L, Maupain C, Monin M-L, Ewenczyk C, Isnard R, Alkouri R, et al. Significance of NT-proBNP and High-Sensitivity Troponin in Friedreich Ataxia. Journal of Clinical Medicine. 2020;9(6):1630.
24. Lynch DR, Sharma S, Hearle P, Greeley N, Gunther K, Keita M, et al. Characterization of clinical serum cardiac biomarker levels in individuals with Friedreich ataxia. Journal of the neurological sciences. 2024;461:123053.
25. Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, Ganiats TG, et al. 2009 Focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines Developed in Collaboration With the International Society for Heart and Lung Transplantation. Journal of the American College of Cardiology. 2009;53(15):e1-e90.
26. GBA. Gemeinsamer Bundesausschuss zu Omaveloxolon bei Friedreich-Ataxie 2024.
27. McDonagh TA, Metra M, Adamo M, Gardner RS, Baumbach A, Böhm M, et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) With the special contribution of the Heart Failure Association (HFA) of the ESC. European heart journal. 2021;42(36):3599-726.
28. Van Gelder IC, Rienstra M, Bunting KV, Casado-Arroyo R, Caso V, Crijns HJGM, et al. 2024 ESC Guidelines for the management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS): Developed by the task force for the management of atrial fibrillation of the European Society of Cardiology (ESC), with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC. Endorsed by the European Stroke Organisation (ESO). European heart journal. 2024;45(36):3314-414.
Bildquellen
Titel: © GAYSORN – stock.adobe.com
Tutorielle Unterstützung
Die tutorielle Unterstützung der Fortbildungsteilnehmer erfolgt durch unseren ärztlichen Leiter Dr. med. Alexander Voigt in Zusammenarbeit mit der arztCME-Redaktion. Inhaltliche Fragen können über das Kommentarfeld, direkt per Mail an service@arztcme.de oder via Telefon unter Tel.: +49(0)180-3000759 gestellt werden. Inhaltliche Fragen werden von unserem ärztlichen Leiter bzw. nach Rücksprache mit diesem und evtl. dem Autor auch von der arztCME-Redaktion beantwortet.
Technischer Support
Der technische Support der Online-Akademie arztCME.de erfolgt durch geschulte Mitarbeiterinnen und Mitarbeiter des Betreibers health&media GmbH unter der E-Mail-Adresse technik@arztcme.de oder via Telefon unter Tel.: 49(0)180-3000759.